
Comparative Efficacy and Tolerability of 15 Antipsychotic Drugs in Schizophrenia: A Multiple-Treatments Meta-Analysis
Abstract
Summary
Background
The question of which antipsychotic drug should be preferred for the treatment of schizophrenia is controversial, and conventional pairwise meta-analyses cannot provide a hierarchy based on the randomised evidence. We aimed to integrate the available evidence to create hierarchies of the comparative efficacy, risk of all-cause discontinuation, and major side-effects of antipsychotic drugs.
Methods
We did a Bayesian-framework, multiple-treatments meta-analysis (which uses both direct and indirect comparisons) of randomised controlled trials to compare 15 antipsychotic drugs and placebo in the acute treatment of schizophrenia. We searched the Cochrane Schizophrenia Group’s specialised register, Medline, Embase, the Cochrane Central Register of Controlled Trials, and ClinicalTrials.gov for reports published up to Sept 1, 2012. Search results were supplemented by reports from the US Food and Drug Administration website and by data requested from pharmaceutical companies. Blinded, randomised controlled trials of patients with schizophrenia or related disorders were eligible. We excluded trials done in patients with predominant negative symptoms, concomitant medical illness, or treatment resistance, and those done in stable patients. Data for seven outcomes were independently extracted by two reviewers. The primary outcome was efficacy, as measured by mean overall change in symptoms. We also examined all-cause discontinuation, weight gain, extrapyramidal side-effects, prolactin increase, QTc prolongation, and sedation.
Findings
We identified 212 suitable trials, with data for 43 049 participants. All drugs were significantly more effective than placebo. The standardised mean differences with 95% credible intervals were: clozapine 0·88, 0·73–1·03; amisulpride 0·66, 0·53–0·78; olanzapine 0·59, 0·53–0·65; risperidone 0·56, 0·50–0·63; paliperidone 0·50, 0·39–0·60; zotepine 0·49, 0·31–0·66; haloperidol 0·45, 0·39–0·51; quetiapine 0·44, 0·35–0·52; aripiprazole 0·43, 0·34–0·52; sertindole 0·39, 0·26–0·52; ziprasidone 0·39, 0·30–0·49; chlorpromazine 0·38, 0·23–0·54; asenapine 0·38, 0·25–0·51; lurasidone 0·33, 0·21–0·45; and iloperidone 0·33, 0·22–0·43. Odds ratios compared with placebo for all-cause discontinuation ranged from 0·43 for the best drug (amisulpride) to 0·80 for the worst drug (haloperidol); for extrapyramidal side-effects 0·30 (clozapine) to 4·76 (haloperidol); and for sedation 1·42 (amisulpride) to 8·82 (clozapine). Standardised mean differences compared with placebo for weight gain varied from −0·09 for the best drug (haloperidol) to −0·74 for the worst drug (olanzapine), for prolactin increase 0·22 (aripiprazole) to −1·30 (paliperidone), and for QTc prolongation 0·10 (lurasidone) to −0·90 (sertindole). Efficacy outcomes did not change substantially after removal of placebo or haloperidol groups, or when dose, percentage of withdrawals, extent of blinding, pharmaceutical industry sponsorship, study duration, chronicity, and year of publication were accounted for in meta-regressions and sensitivity analyses.
Interpretation
Antipsychotics differed substantially in side-effects, and small but robust differences were seen in efficacy. Our findings challenge the straightforward classification of antipsychotics into first-generation and second-generation groupings. Rather, hierarchies in the different domains should help clinicians to adapt the choice of antipsychotic drug to the needs of individual patients. These findings should be considered by mental health policy makers and in the revision of clinical practice guidelines.
(Reprinted with permission from Lancet 2013; 382: 951–62)
Introduction
Schizophrenia is a debilitating disease, ranked among the top 20 causes of disability worldwide.1 The question of which antipsychotic drug should be preferred for treatment of the disease is controversial, largely because of the substantial costs of second-generation antipsychotic drugs (estimated US$14·5 billion globally in 2014).2 New antipsychotic drugs such as asenapine, iloperidone, lurasidone, and paliperidone continue to be marketed, but as earlier second-generation drugs come off patent, an important question is whether the newest drugs are cost effective. Previous conventional pairwise meta-analyses3–5 could not generate clear hierarchies for the efficacy and side-effects of available treatments, because many antipsychotic drugs have not been compared head to head,6 and because such analyses could not integrate all the evidence from several comparators. As such, any attempt to create such hierarchies was necessarily impressionistic, and guidelines urgently need accurate information to address this question. We aimed to compare the two prototypal first-generation (haloperidol and chlorpromazine) and 13 second-generation anti-psychotic drugs when used in patients with schizophrenia. Our intention was to provide evidence-based hierarchies of the comparative efficacy, risk of all-cause discontinuation, and major side-effects of antipsychotic drugs.
Methods
Participants and Interventions
We did a multiple-treatments meta-analysis to compare 15 antipsychotic drugs for schizophrenia. Multiple-treatments meta-analysis allows the integration of direct and indirect comparisons of antipsychotic drugs (ie, how two or more drugs compare with a common comparator). We followed the same approach as was used in two previous multiple-treatments meta-analyses, of major depressive disorder7 and bipolar mania.8
Our analysis included studies of people with schizophrenia or related disorders (schizoaffective, schizophreniform, or delusional disorder [as defined by any diagnostic criteria]). Because multiple-treatments meta-analysis requires a reasonably homogeneous sample,9,10 we excluded randomised controlled trials done in patients with predominant negative symptoms, concomitant medical illness, or treatment resistance, and trials in patients with stable illness (mainly relapse prevention studies).
We included studies of 15 orally administered antipsychotic drugs used as monotherapies, including all flexible-dose studies since these allow the investigators to titrate to the adequate dose for the individual patient. For fixed-dose studies, we included target doses up to maximum doses on the basis of those established by the international consensus study of antipsychotic dosing,11 which are justified by available evidence and are similar to other recommendations12–14 (appendix pp 25–40). Only 40 out of 474 (8%) active study arms were excluded on this basis and not addressed in a sensitivity analysis (appendix pp 41–65), and dose was addressed by several meta-regression and sensitivity analyses.
Search Strategy and Selection Criteria
We started by collating the reports identified in seven previous systematic reviews.3,6,15–19 We then searched the Cochrane Schizophrenia Group’s specialised register (compiled by regular systematic searches of numerous databases, clinical trial registers, hand searches, and conference proceedings20 available up to August, 2009), Medline, Embase, PsycINFO, the Cochrane Central Register of Controlled Trials, and ClinicalTrials.gov for reports published up to Sept 1, 2012. Search terms were the generic names of the antipsychotic drugs as well as QT*, electrocard*, arrhythm*, ecg, and prolactin* (appendix pp 70–76). We also checked relevant reports on the US Food and Drug Administration (FDA) website, checked the references lists of other reviews,21,22 and searched the websites of pharmaceutical companies, which were also asked to provide additional information about their studies.
We included published and unpublished randomised controlled trials that were at least single-blinded in our analysis. Studies in which sequence generation had a high risk of bias or in which allocation was clearly not concealed (eg, alternate allocation) were excluded. Unblinded studies were excluded because they systematically favoured second-generation drugs in a previous analysis.3 We decided a priori to exclude studies from China to avoid a systematic bias, since many of these studies do not use appropriate randomisation procedures and do not report their methods.23 We also excluded trials that allowed switching between groups. Study quality was independently assessed by two of five reviewers (FR, DÖ, SL, MPS, BL), who used the Cochrane Collaboration’s risk-of-bias method.24
Outcome Measures and Data Extraction
The primary outcome was the mean overall change in symptoms, which was assessed in the first instance by change in Positive and Negative Syndrome Scale25 (total score from baseline to endpoint); if data from this scale were not available, we used change in Brief Psychiatric Rating Scale26 from baseline to endpoint, and then values at study endpoint of these scales. Intention-to-treat datasets were used whenever available. Secondary outcomes were all-cause discontinuation, weight gain, use of antiparkinson drugs as a measure of extrapyramidal side-effects, prolactin increase, QTc prolongation, and sedation. Studies in which antiparkinson drugs were given prophylactically were excluded from the analysis of extrapyramidal side-effects. Because multiple-treatments meta-analysis requires reasonable homogeneity we focused on acute treatment, which we defined as 6-weeks duration. If 6-week data were not available, we used data from between 4 and 12 weeks (the datapoint closest to 6 weeks was given preference).
Study selection and data extraction were done independently by at least two of eight reviewers (FR, DÖ, SL, LS, AC, MS, MPS, and BL). Data extraction forms were sent to original authors of trial reports when necessary with a request to provide missing data and the option to make corrections. Missing standard deviations were estimated from p values or with the mean standard deviation of the other included studies.27
Statistical Analysis
Multiple-treatments meta-analysis combines direct and indirect evidence for all relative treatment effects and provides estimates with maximum power.28–31 The model was fitted into a Bayesian context with hierarchical models (appendix pp 66–69). A common heterogeneity parameter was assumed for all comparisons. For continuous outcomes, the relative effect sizes were calculated as standardised mean differences (Hedges’ g). For binary outcomes, relative effect sizes were calculated as odds ratios (ORs). Both types of effect sizes are reported with their 95% credible intervals (CrIs). To rank the treatments we used the surface under the cumulative ranking (SUCRA) probabilities.31 SUCRAs expressed as percentages compare each intervention to an imaginary intervention that is always the best without uncertainty.
A SUCRA of x% means that the drug achieves x% of the effectiveness of this imaginary drug, thus larger SUCRAs denote more effective interventions. Numbers needed to treat (NNT) and numbers needed to harm (NNH) were estimated with the average occurrence of an outcome as the baseline risk.
The underlying assumption of transitivity suggests that all pairwise comparisons in the network do not differ with respect to the distribution of effect modifiers.32 Inconsistency between direct and indirect evidence would suggest transitivity is not apparent between results.9,10 Consistency was mainly assessed by the comparison of the conventional network meta-analysis model, for which consistency is assumed, with a model that does not assume consistency (a series of pairwise meta-analyses analysed jointly). If the trade-off between model fit and complexity favoured the model with assumed consistency, this model was preferred (appendix pp 66–69).33 Moreover, we calculated the difference between direct and indirect evidence in all closed loops in the network; inconsistent loops were identified with a significant (95% CrI that excludes 0) disagreement between direct and indirect evidence.34 A loop of evidence is a collection of studies that links treatments to allow for indirect comparisons; the simplest loop is a triangle formed by three direct comparison studies with shared comparators.
We did several sensitivity analyses on the primary outcome to explore potential reasons for heterogeneity or inconsistency. Those planned in advance were exclusion of: studies that compared high doses of one drug with low doses of the other (defined a priori in the protocol [appendix pp 2–24]; n=5); single-blinded studies (n=7); and first-episode studies (n=7). Other analyses were post hoc: inclusion of some previously excluded fixed dose groups on the basis of the FDA rule (more effective than placebo in a least two trials; appendix pp 25–40); exclusion of haloperidol to rule out differences in its dose as a potential bias (n=54);35 exclusion of placebo since reduced efficacy of newer drugs could be due to increasing placebo response (n=43);36 exclusion of studies with missing standard deviations (n=19); exclusion of studies that were not analysed on an intention-to-treat basis (n=18); and exclusion of so-called failed studies (in which both the new drug and the active comparator were not more effective than placebo; n=6).
Post-hoc multiple-treatments meta-regression was used to examine the effects of unfair dose comparisons (as independently judged by SL and JD). In another analysis we classified haloperidol treatment groups into those in which patients received doses of 12 mg per day or less and those in which they received more than 12 mg per day (a cutoff that showed a significant dose effect in a previous meta-analysis4) and into those in which they received 7·5 mg per day or less, or more than 7·5 mg per day (on the basis of a Cochrane review37); classified chlorpromazine treatment groups into those in which patients received doses of 600 (or 500) mg per day or less, or more than 600 (or 500) mg per day (to replicate the cutoff used by Leucht and colleagues38); and used the difference in dose expressed by olanzapine equivalents (on the basis of the international consensus study of antipsychotic dosing.11 Because asenapine, iloperidone, and lurasidone were not included in the international consensus study,11 we assumed that their maximum label dose corresponded to olanzapine at 20 mg per day, because the investigators of that study had made similar decisions for most other drugs.
Other preplanned meta-regressions addressed sponsorship (whether the sponsor was the manufacturer of the test or comparator drug), the mean age of trial participants (used as a proxy for chronicity, because mean duration of illness was inconsistently reported), year of publication, study duration, and overall percentage of withdrawals. A post-hoc subgroup analysis compared the results of trials reported up to the end of 1997 and those reported after 1997. All analyses related to dose were also done for the outcome of extrapyramidal side-effects (as measured by use of antiparkinson drugs). We explored small-study effects in the placebo-controlled trials with a funnel-plot technique expanded to multiple-treatments meta-analysis and accounted for such effects via network meta-regression with the standard error as covariate.39,40
Our study protocol was made freely available to the public on two of our institutional websites, and is included in the appendix (pp 2–24).
Role of the Funding Source
No specific funding was received for this work. GS and LS were supported by a grant from the European Research Council (IMMA 260559). These funders had no role in study design, data collection, analysis or interpretation, or writing of the report.
Results
212 studies reported between October, 1955, and September, 2012, with 43049 participants, were included in the analysis (details of included studies are shown in appendix pp 41–65; PRISMA41 flowcharts are shown in appendix pp 70–76). The mean duration of illness was 12·4 years (SD 6·6) and the mean age of trial participants was 38·4 years (SD 6·9). Nine studies exclusively examined first-episode patients. In terms of study quality, the reports often did not provide details about randomisation procedures and allocation concealment (appendix pp 77–84); however, 144 studies (68%) were done by pharmaceutical companies, which (in those cases in which they responded to our requests for information) had used appropriate methods throughout. 13 studies were single-blinded (with allocation concealed from assessors), and the rest (199, 94%) were double-blinded, but few details were reported about the methods of concealment or how successful they were. Our analysis accorded with the known high numbers of withdrawals in clinical studies of schizophrenia (35% overall for the studies included in our analysis); the effect of withdrawals was examined by meta-regression. The main reason for selective reporting was that the use of antiparkinson drugs was often not reported. The networks of eligible comparisons are shown in figure 1 and in the appendix (pp 85–91). The results of the direct comparisons for all outcomes are shown in the appendix (pp 92–96).
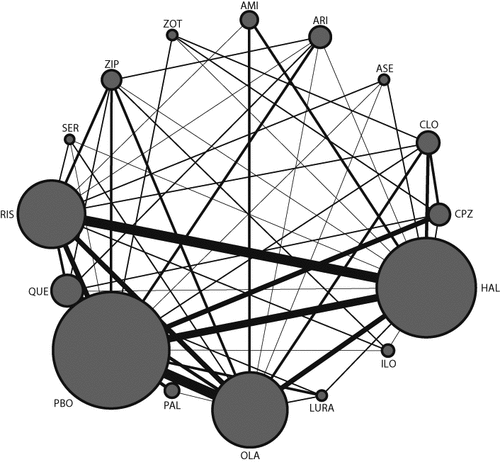
The size of the nodes corresponds to the number of trials that study the treatments. Directly comparable treatments are linked with a line, the thickness of which corresponds to the number of trials that assess the comparison. AMI=amisulpride. ARI=aripiprazole. ASE=asenapine. CLO=clozapine. CPZ=chlorpromazine. HAL=haloperidol. ILO=iloperidone. LURA=lurasidone. OLA=olanzapine. PAL=paliperidone. PBO=placebo. QUE=quetiapine. RIS=risperidone. SER=sertindole. ZIP=ziprasidone. ZOT=zotepine.
We created hierarchies of effect size on the basis of SUCRA rankings for all outcomes. Figures 2 and 3 show these results for overall efficacy (appendix pp 97–104). Most of the differences between drugs are gradual rather than discrete. As a rule of thumb, Cohen42 has suggested that a standardised mean difference of −0·2 is small, −0·5 medium, and −0·8 large. All drugs were superior to placebo (range of mean effect sizes −0·33 to −0·88; figure 3), and clozapine was significantly more effective than all the other drugs (figure 2). After clozapine, amisulpride, olanzapine, and risperidone were significantly more effective than the other drugs apart from paliperidone and zotepine. These effect sizes were small (range −0·11 to −0·33; figure 2).
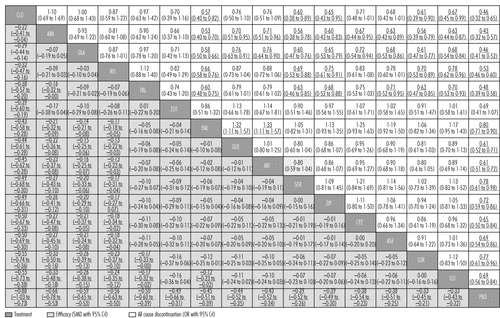
Drugs are reported in order of efficacy ranking. Comparisons between treatments should be read from left to right and the estimate is in the cell in common between the column-defining treatment and the row-defining treatment. For efficacy, standard mean differences (SMDs) lower than 0 favour the column-defining treatment. For all-cause discontinuation, odds ratios (ORs) higher than 1 favour the column-defining treatment. To obtain SMDs for comparisons in the opposite direction, negative values should be converted into positive values, and vice versa. To obtain ORs for comparisons in the opposite direction, reciprocals should be taken. Significant results are in bold and underlined. CLO=clozapine. AMI=amisulpride. OLA=olanzapine. RIS=risperidone. PAL=paliperidone. ZOT=zotepine. HAL=haloperidol. QUE=quetiapine. ARI=aripiprazole. SER=sertindole. ZIP=ziprasidone. CPZ=chlorpromazine. ASE=asenapine. LUR=lurasidone. ILO=iloperidone. PBO=placebo.
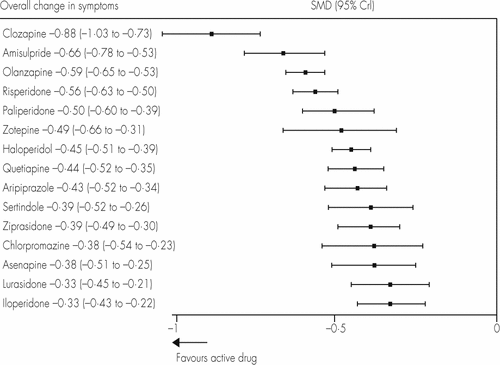
Treatments are ranked according to their surface under the cumulative ranking (SUCRA) values (appendix p 98). SMD=standardised mean difference. CrI=credible interval.
All-cause discontinuation was used as a measure of acceptability. All drugs were significantly better than placebo apart from zotepine (figure 2, figure 4A). ORs and NNTs ranged from 0·43 and 6 for amisulpride to 0·80 and 20 for haloperidol. Amisulpride (range of significant mean ORs 0·53–0·71; NNTs 8–14), olanzapine (0·58–0·76; 9–17), clozapine (0·57–0·67; 9–12), paliperidone (0·60–0·71; 9–14), and risperidone (0·66–0·78; 11–18) had significantly lower all-cause discontinuation than several other drugs. Haloperidol was worse than quetiapine (OR 1·32; NNT 15) and aripiprazole (OR 1·33; NNT 15; figure 2; for NNTs and NNHs see appendix pp 133–39).
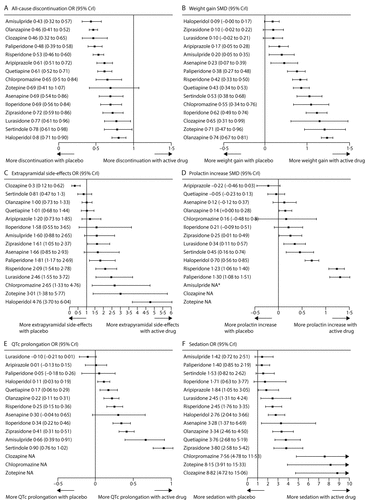
Results are shown for all-cause discontinuation (A), weight gain (B), extrapyramidal side-effects (C), prolactin increase (D), QTc prolongation (E), and sedation (F). Treatments are ranked according to their surface under the cumulative ranking (SUCRA) values (appendix pp 97–104). Extrapyramidal side-effects are defined by at least one use of antiparkinson drugs. OR=odds ratio. CrI=credible interval. SMD=standardised mean difference. *In one small study,43 amisulpride (mean 473 mg per day) produced less prolactin increase than haloperidol (mean 28 mg per day), but prolactin concentrations were highly imbalanced at baseline, so we excluded this result (inclusion of this study in the analysis did not affect the ranking of the other drugs).
Apart from haloperidol, ziprasidone, and lurasidone, all drugs produced more weight gain than placebo (figures 4B, 5). Olanzapine produced significantly more weight gain than most other drugs, followed by zotepine (figure 5). Clozapine, iloperidone, chlorpromazine, sertindole, quetiapine, risperidone, and paliperidone produced significantly more weight gain than haloperidol, ziprasidone, lurasidone, aripiprazole, amisulpride, and asenapine (with the exception that asenapine did not differ significantly from paliperidone). Standardised mean differences for these comparisons ranged from −0·18 to −0·57 (figure 5). Other differences were not statistically significant apart from iloperidone causing more weight gain than paliperidone, risperidone, and quetiapine (figure 5).
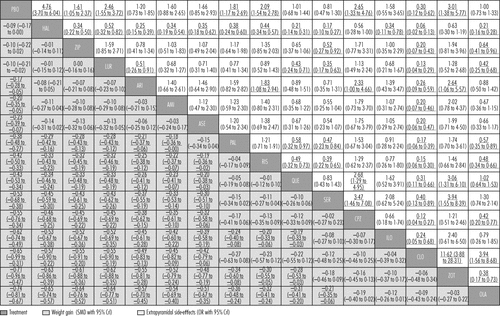
Drugs are reported in order of weight-gain ranking. Comparisons between treatments should be read from left to right and the estimate is in the cell in common between the column-defining treatment and the row-defining treatment. For weight gain, standard mean differences (SMDs) lower than 0 favour the column-defining treatment. For movement disorders, odds ratios (ORs) higher than 1 favour the row-defining treatment.To obtain SMDs for comparisons in the opposite direction, negative values should be converted into positive values, and vice versa. To obtain ORs for comparisons in the opposite direction, reciprocals should be taken. Significant results are in bold and underlined. Extrapyramidal side-effects are defined by at least one use of antiparkinson drugs. PBO=placebo. HAL=haloperidol. ZIP=ziprasidone. LUR=lurasidone. ARI=aripiprazole. AMI=amisulpride. ASE=asenapine. PAL=paliperidone. RIS=risperidone. QUE=quetiapine.
Clozapine, sertindole, olanzapine, quetiapine, aripiprazole, iloperidone, amisulpride, and asenapine did not cause significantly more extrapyramidal side-effects than placebo. The range of mean ORs and NNHs for the other drugs were 1·61–4·76 and 3–11, respectively (figure 4C). Clozapine produced fewer extrapyramidal side-effects than all other drugs and placebo (mean ORs 0·06–0·40; NNTs 5–9), and was followed in ranking by sertindole, olanzapine, and quetiapine (figure 5, for NNTs see appendix pp 133–39). Haloperidol caused significantly more extrapyramidal side-effects than the other drugs apart from zotepine and chlorpromazine, for which the differences were not significant (mean ORs 0·06–0·52; NNHs 5–11; in favour of other drugs). Zotepine, chlorpromazine, lurasidone, risperidone, and paliperidone were among the least well tolerated drugs, because they produced significantly more extrapyramidal side-effects than several others in the analysis (figure 5).
Aripiprazole, quetiapine, asenapine, chlorpromazine, and iloperidone did not cause significantly increased prolactin concentrations compared with placebo (figure 4D). Paliperidone and risperidone were associated with significantly more prolactin increase than all other drugs including haloperidol, and haloperidol was associated with significantly more than the rest apart from chlorpromazine and sertindole (figure 6). Clozapine and zotepine could not be included in the analysis, because the one direct comparison between them (ie, with each other) was not linked with any other drug in the network (standardised mean difference −1·23, 95% CrI −1·8 to −0·64, in favour of clozapine; n=52).44 No usable data were available for amisulpride.
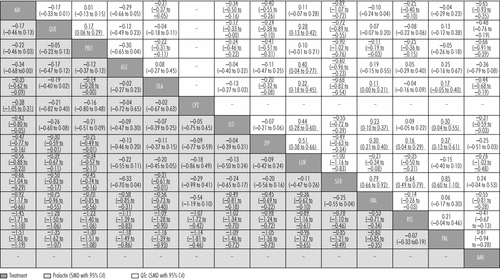
Drugs are reported in order of prolactin increase ranking. Comparisons between treatments should be read from left to right and the estimate is in the cell in common between the column-defining treatment and the row-defining treatment. For prolactin increase, standard mean differences (SMDs) lower than 0 favour the column-defining treatment. For QTc prolongation, SMDs lower than 0 favour the row-defining treatment. To obtain SMDs for comparisons in the opposite direction, negative values should be converted into positive values, and vice versa. Significant results are in bold and underlined. Clozapine and zotepine could not be included in the analysis, because their only comparison with each other was not linked with any other drug in the network. ARI=aripiprazole. QUE=quetiapine. PBO=placebo. ASE=asenapine. OLA=olanzapine. CPZ=chlorpromazine. ILO=iloperidone. ZIP=ziprasidone. LUR=lurasidone. SER=sertindole. HAL=haloperidol. RIS=risperidone. PAL=paliperidone. AMI=amisulpride.
Lurasidone, aripiprazole, paliperidone, and asenapine were not associated with significant QTc prolongation compared with placebo (figure 4E). The standardised mean differences of the other drugs compared with placebo ranged from marginal (0·11, haloperidol) to large (0·90, sertindole). Results for the comparisons between drugs with respect to QTc prolongation are shown in figure 6.
Amisulpride, paliperidone, sertindole, and iloperidone were not significantly more sedating than placebo (figure 4F). For the other drugs compared with placebo, mean ORs and NNHs ranged from 1·84 and 10 (aripiprazole) to 8·82 and 2 (clozapine). ORs for the comparisons between drugs with respect to sedation are shown in figure 7 and their NNTs are shown in the appendix (pp 133–39).

Drugs are reported in order of sedation ranking. Comparisons between treatments should be read from left to right and the estimate is in the cell in common between the column-defining treatment and the row-defining treatment. For sedation, odds ratios (ORs) higher than 1 favour the column-defining treatment. To obtain ORs for comparisons in the opposite direction, reciprocals should be taken. Significant results are in bold and underlined. PBO=placebo. AMI=amisulpride. PAL=paliperidone. SER=sertindole. ILO=iloperidone. ARI=aripiprazole. LUR=lurasidone. RIS=risperidone. HAL=haloperidol. ASE=asenapine. OLA=olanzapine. QUE=quetiapine. ZIP=ziprasidone. CPZ=chlorpromazine. ZOT=zotepine. CLO=clozapine.
The assumption of consistency was generally supported by a better trade-off between model fit and complexity when consistency was assumed than when it was not (appendix pp 105–14). Significant disagreement between direct and indirect estimates (inconsistency) was identified in only very few cases: for efficacy seven of 80 loops; for all-cause discontinuation three of 80 loops; for weight gain one of 62 loops; for extrapyramidal side-effects one of 56 loops; for prolactin increase three of 44 loops; for QTc prolongation two of 35 loops; and for sedation none of 49 loops were inconsistent (appendix pp 105–14). Data were double-checked and we could not identify any important variable that differed across comparisons in these loops. The number of included studies in the inconsistent loops was typically small, so the extent of inconsistency was not substantial enough to change the results.
Results for efficacy and extrapyramidal side-effects were robust against the sensitivity and meta-regression analyses (appendix pp 115–32). The most notable exceptions were that the relative efficacy of asenapine increased from the 13th to the seventh rank when placebo comparisons were removed. A large, failed study had driven its primary result, so asenapine was also more effective (ninth rank) when such trials were excluded. Haloperidol doses lower than 12 mg per day (or 7·5 mg per day) caused significantly fewer extrapyramidal side-effects than did higher doses, but still more than any other antipsychotic drug; for the efficacy outcome, lower doses of haloperidol did not significantly differ from higher doses. Chlorpromazine doses higher than 600 mg per day (or 500 mg per day) were associated with higher efficacy (sixth rank) than lower doses (14th rank), with little difference in extrapyramidal side-effects. Small studies tended to show higher efficacy of the active interventions compared with placebo (regression coefficient=1·31, 95% CrI 0·58–2·03). However this had only a small effect on the ranking of the treatments (appendix pp 115–32). None of the other meta-regression or sensitivity analyses led to any important changes in the efficacy and extrapyramidal side-effect hierarchies (appendix pp 115–32).
Discussion
Our multiple-treatments meta-analysis provides evidence-based hierarchies for the efficacy and tolerability of antipsychotic drugs, overcoming the major limitation of conventional pairwise meta-analyses.3,4,21 Results for our primary outcome challenge the dogma that the efficacy of all antipsychotic drugs is the same. This notion originated from an influential narrative review published in 1969,45 but it has not been scientifically addressed since.
The efficacy hierarchy generated by our analysis was robust against many sources of bias, including various analyses related to dose. In particular, findings from pairwise meta-analyses3–5 suggested that some, but not all, second-generation antipsychotics were more effective than haloperidol, but these findings have been criticised for differences in haloperidol doses used by the included studies, which might have affected the efficacy outcomes.35 However, the fact that exclusion of all haloperidol comparisons in our analysis did not affect the efficacy hierarchy refutes this criticism. The FDA still requires placebo-controlled trials for all new antipsychotic drugs. Increasing placebo response in such trials is a concern,36 but exclusion of all placebo comparisons did not change the results much in our analysis, apart from asenapine turning out more effective than in the primary analysis. That the four most effective second-generation antipsychotic drugs were the first to be developed could also suggest a cohort effect in terms of changes in study populations. However, two meta-regression analyses— one with publication year as a continuous moderator and the other comparing the results of trials published in the past 15 years with those published earlier—did not change the efficacy hierarchy to an important extent. The example of paliperidone (approved by the FDA in 2007), which is the active metabolite of risperidone (approved by the FDA in 1993), and has essentially the same receptor-binding profile,46 also contradicts this suggestion, because both drugs ranked next to each other in most domains (apart from sedation and QTc prolongation) and because paliperidone was more effective than several antipsychotic drugs that had been developed previously (figures 2, 3).
We emphasise that the differences in efficacy between drugs were small (standardised mean differences 0·11–0·55, median 0·24), and smaller overall than those for side-effects. However, for perspective, the efficacy differences compared with placebo were of only medium size (0·33–0·88, median 0·44), so the differences in efficacy between drugs are possibly substantial enough to be clinically important. Finally, because most clozapine studies were done in refractory patients, clozapine is thought to be superior only in this subtype, but in our analysis of non-refractory patients it was also more effective than all the other drugs. However, this result has the limitation that it was mainly based on older comparisons of clozapine with first-generation drugs. As in our previous conventional meta-analysis,47 clozapine was not more effective than any other second-generation antipsychotic in direct pairwise comparisons (appendix pp 92–96). A European Union-funded study to examine the early use of clozapine in first-episode patients is underway.
All-cause discontinuation has previously been used as a measure for the acceptability of treatments, because it encompasses efficacy and tolerability.7,8 In our analysis, the results paralleled the efficacy findings in that the most effective drugs also had the lowest discontinuation rates (although haloperidol, the worst drug with respect to all-cause discontinuation, had a middle rank for efficacy). In randomised controlled trials in patients with schizophrenia, more participants withdraw because of inefficacy (40% overall for the studies included in our analysis) than because of side-effects (17%; other reasons for withdrawal were not assessed),36 and some evidence suggests that patients prioritise efficacy over tolerability.37 We have used the neutral term all-cause discontinuation, because clinicians might intuitively associate the word acceptability more with tolerability than with efficacy.
Haloperidol caused the most extrapyramidal side-effects, followed by zotepine and chlorpromazine. Chlorpromazine did not produce significantly more extrapyramidal side-effects than did most second-generation antipsychotics. Haloperidol doses lower than 7·5 mg per day (the lowest dose in multiple-episode patients was 4 mg per day) produced similar outcomes for efficacy and extrapyramidal side-effects as did higher doses. However, five second-generation drugs were associated with significantly more extrapyramidal side-effects than was placebo. These findings show that extrapyramidal side-effects cannot be used for a dichotomous classification into first-generation and second-generation antipsychotics. Curiously, clozapine was associated with less use of antiparkinson drugs than was placebo. Abrupt withdrawal of prestudy treatment and too short washout phases (sometimes only 48 h) can lead to rebound and carry-over extrapyramidal side-effects.48 Furthermore, involuntary movements are present in 9–17% of antipsychotic drug-naive people with schizophrenia.49 Clozapine has a low intrinsic risk of extrapyramidal side-effects and might suppress both of these effects.
Weight gain and associated metabolic problems are regarded as the major issues associated with new antipsychotic drugs. Indeed, olanzapine, zotepine, and clozapine were the worst in this respect, and some guidelines recommend against the first-line use of olanzapine for first-episode patients.12 However, ziprasidone and lurasidone (along with haloperidol) were the only antipsychotic drugs without significantly more weight gain than placebo in adults. By contrast, chlorpromazine was among the worst drugs in this respect. This finding shows that sedating, low-potency, first-generation antipsychotics also cause weight gain, and that a dichotomy between first-generation and second-generation antipsychotics based on weight gain is another oversimplification.3
Sedation is unpleasant for patients. Overall, our results with respect to sedation were reasonable, and direct and indirect comparisons were consistent. For example, clozapine and chlorpromazine are certainly sedating drugs; the good results for amisulpride can be accounted for by the absence of blockade of histaminergic receptors associated with sedation; and the small sedative effects of paliperidone can possibly be accounted for by its slow-release mechanism limiting plasma peaks after ingestion. Although the highest ORs were almost two-times higher for sedation than the highest for extrapyramidal side-effects, sedation is sometimes transient, is measured only by spontaneous reports, and the potential confounder of concomitant use of benzodiazepines in the studies should not be ignored.
QTc prolongation can lead to life-threatening torsades de pointes.50 The antipsychotic drugs assessed differed enormously with respect to this outcome, with some not differing from placebo, and one (sertindole) being almost one standard deviation worse. Indeed, sertindole was associated with increased cardiac mortality compared with risperidone in a large, pragmatic, randomised controlled trial51 (n=9858, all-cause mortality not different). In another study,52 no difference in frequency of sudden death was seen between ziprasidone (the third worst drug in our analysis) and olanzapine (n=18154).51 We emphasise that amisulpride was regarded as benign in some guidelines,13 but our findings show that it might not be—a result that is consistent with an analysis of amisulpride overdoses.50 This result has the limitation that the evidence is indirectly derived from two comparisons with olanzapine, since direct comparisons with placebo were not available (appendix pp 92–96). QTc data were not available for the older drugs (clozapine, chlorpromazine, and zotepine).
Prolactin increase can be associated with several side-effects such as amenorrhoea, galactorrhoea, sexual dysfunction, and osteoporosis; a possible association with breast cancer has also been discussed, but the link is not proven.53 The causes of some of these side-effects are multifactorial—eg, decreased libido can also be the expression of schizophrenic negative symptoms, and osteoporosis can be caused by immobility in schizophrenia. However, the differences between drugs with respect to this outcome were large. For example, paliperidone and risperidone increased prolactin by more than one standard deviation compared with placebo; aripiprazole reduced prolactin (although not significantly) because of its partial-dopamine-agonist properties. Despite the collaboration of its manufacturer, no useable data on amisulpride were available, but its high prolactin risk is well known.54
Our study has several limitations. The network could be expanded to old drugs such as perphenazine and sulpiride, which have had good results in effectiveness studies,55,56 but only a few relevant perphenazine trials have been done.57 As more and more second-generation antipsychotics are losing their patent protection, the debate about the costs of the original second-generation antipsychotics becomes less important. The present debate is about whether the newest drugs are cost-effective. These new drugs do have favourable properties, such as acceptable weight gain (especially lurasidone and asenapine, and to a lesser extent iloperidone and paliperidone). De Hert and colleagues22,58 additionally noted that these drugs might be fairly benign with respect to increases of lipids and glucose, which partly correspond to weight gain. Reporting of side-effects is unsatisfactory in randomised controlled trials in patients with psychiatric disorders,59 and some side-effects were not recorded at all for some drugs (figure 4). The meta-regression with percentage of withdrawals as a moderator could not rule out all potential bias associated with high attrition in schizophrenia trials.
Our findings cannot be generalised to young people with schizophrenia, patients with predominant negative symptoms, refractory patients, or stable patients, all of whom were excluded to enhance homogeneity as required by multiple-treatments meta-analysis. A funnel-plot asymmetry was seen, which is not necessarily the expression of publication bias, but rather of higher efficacy in small trials than in larger ones, for various reasons.24 For example, sample size estimates for drugs with low efficacy might have needed higher numbers of participants to attain statistical significance than in trials with more effective drugs. However, accounting for trial size did not substantially change the rankings. Finally, because multiple-treatments meta-analysis requires reasonably homogeneous studies, we had to restrict ourselves to short-term trials. Because schizophrenia is often a chronic disorder, future multiple-treatments meta-analyses could focus on long-term trials,60 but these remain scarce.3 In any case, for clinicians to know to which drugs patients are most likely to respond within a reasonable duration such as 6 weeks is important.
Antipsychotic drugs differ in many properties and can therefore not be categorised in first-generation and second-generation groupings. The suggested hierarchies in seven major domains should help clinicians to adapt choice of antipsychotic drug to the needs of individual patients, and should lead to modification of clinical practice guidelines.
1 . Years lived with disability (YLDs) for 1160 sequelae of 289 diseases and injuries 1990-2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet 2012; 380: 2163–96.Crossref, Google Scholar
2
3 . Second-generation versus first-generation antipsychotic drugs for schizophrenia: a meta-analysis. Lancet 2009; 373: 31–41.Crossref, Google Scholar
4 . Atypical antipsychotics in the treatment of schizophrenia: systematic overview and meta-regression analysis. BMJ 2000; 321: 1371–76.Crossref, Google Scholar
5 . A meta-analysis of the efficacy of second-generation antipsychotics. Arch Gen Psychiatry 2003; 60: 553–64.Crossref, Google Scholar
6 . A meta-analysis of head to head comparisons of second generation antipsychotics in the treatment of schizophrenia. Am J Psychiatry 2009; 166: 152–63.Crossref, Google Scholar
7 . Comparative efficacy and acceptability of 12 new-generation antidepressants: a multiple-treatments meta-analysis. Lancet 2009; 373: 746–58.Crossref, Google Scholar
8 . Comparative efficacy and acceptability of antimanic drugs in acute mania: a multiple-treatments meta-analysis. Lancet 2011; 378: 1306–15.Crossref, Google Scholar
9 . Validity of indirect comparisons in meta-analysis. Lancet 2007; 369: 270.Crossref, Google Scholar
10 . Inconsistency between direct and indirect comparisons of competing interventions: meta-epidemiological study. BMJ 2011; 343: d4909.Google Scholar
11 . International consensus study of antipsychotic dosing. Am J Psychiatry 2010; 167: 686–93.Crossref, Google Scholar
12 . The 2009 schizophrenia PORT psychopharmacological treatment recommendations and summary statements. Schizophr Bull 2010; 36: 71–93.Crossref, Google Scholar
13 . World Federation of Societies of Biological Psychiatry (WFSBP) Guidelines for Biological Treatment of Schizophrenia, part 1: update 2012 on the acute treatment of schizophrenia and the management of treatment resistance. World J Biol Psychiatry 2012; 13: 318–78.Crossref, Google Scholar
14 NICE. Schizophrenia: core interventions in the treatment and management of schizophrenia in primary and secondary care (update). London: National Institute for Health and Clinical Excellence, 2009.Google Scholar
15 . How effective are second-generation antipsychotic drugs? A meta-analysis of placebo-controlled trials. Mol Psychiatry 2009; 14: 429–47.Crossref, Google Scholar
16 Leucht C, Kitzmantel M, Chua L, Kane J, Leucht S. Haloperidol versus chlorpromazine for schizophrenia. Cochrane Database Syst Rev 1: CD004278.Google Scholar
17 Nussbaum A, Stroup TS. Paliperidone for schizophrenia. Cochrane Database Syst Rev 6: CD006369.Google Scholar
18 . Haloperidol versus placebo for schizophrenia. Cochrane Database of Syst Rev 2006; 4: CD003082.Crossref, Google Scholar
19 . Chlorpromazine versus placebo for schizophrenia. Cochrane Database Syst Rev 2007; 2: CD000284.Crossref, Google Scholar
20
21 . Antipsychotics in adults with schizophrenia: comparative effectiveness of first-generation versus second- generation medications: a systematic review and meta-analysis. Ann Intern Med 2012; 157: 498–511.Crossref, Google Scholar
22 . Body weight and metabolic adverse effects of asenapine, iloperidone, lurasidone and paliperidone in the treatment of schizophrenia and bipolar disorder: a systematic review and exploratory meta-analysis. CNS Drugs 2012; 26: 733–59.Crossref, Google Scholar
23 Wu TX, Li YP, Liu GJ, et al. Investigation of authenticity of ‘claimed’ randomized controlled trials (RCTs) and quality assessment of RCT reports published in China. XIV Cochrane Colloquium; Dublin; Oct 23-26, 2006.Google Scholar
24 . Cochrane handbook for systematic reviews of interventions, version 5.1.0. Chichester: John Wiley & Sons, 2011.Google Scholar
25 . The positive and negative symptome scale (PANSS) for schizophrenia. Schizophr Bull 1987; 13: 261–75.Crossref, Google Scholar
26 . The Brief Psychiatric Rating Scale. Psychol Rep 1962; 10: 790–812.Crossref, Google Scholar
27 . Imputing missing standard deviations in meta-analyses can provide accurate results. J Clin Epidemiol 2006; 59: 7–10.Crossref, Google Scholar
28 . Combination of direct and indirect evidence in mixed treatment comparisons. Stat Med 2004; 23: 3105–24.Crossref, Google Scholar
29 . Evaluation of networks of randomized trials. Stat Methods Med Res 2008; 17: 279–301.Crossref, Google Scholar
30 . Borrowing strength from external trials in a meta-analysis. Stat Med 1996; 15: 2733–49.Crossref, Google Scholar
31 . Graphical methods and numerical summaries for presenting results from multiple-treatment meta-analysis: an overview and tutorial. J Clin Epidemiol 2011; 64: 163–71.Crossref, Google Scholar
32 . Indirect and mixed-treatment comparison, network, or multiple-treatments meta-analysis: many names, many benefits, many concerns for the next generation evidence synthesis tool. Res Synth Methods 2012; 3: 80–97.Crossref, Google Scholar
33 . Checking consistency in mixed treatment comparison meta-analysis. Stat Med 2010; 29: 932–44.Crossref, Google Scholar
34 . A case study of multiple-treatments meta-analysis demonstrates that covariates should be considered. J Clin Epidemiol 2009; 62: 857–64.Crossref, Google Scholar
35 . Comparative efficacy of antipsychotics in the treatment of schizophrenia: a critical assessment. Schizophr Res 2005; 79: 145–55.Crossref, Google Scholar
36 . Exploratory analyses of efficacy data from schizophrenia trials in support of new drug applications submitted to the US Food and Drug Administration. J Clin Psychiatry 2012; 73: 856–64.Crossref, Google Scholar
37 . Haloperidol dose for the acute phase of schizophrenia. Cochrane Database Syst Rev 2002; 2: CD001951.Crossref, Google Scholar
38 . New generation antipsychotics versus low-potency conventional antipsychotics: a systematic review and meta-analysis. Lancet 2003; 361: 1581–89.Crossref, Google Scholar
39 . Using network meta-analysis to evaluate the existence of small-study effects in a network of interventions. Res Synth Methods 2012; 3:161–76.Crossref, Google Scholar
40 . Assessment of regression-based methods to adjust for publication bias through a comprehensive simulation study. BMC Med Res Methodol 2009; 9: 2.Crossref, Google Scholar
41 . The PRISMA statement for reporting systematic reviews and meta-analyses of studies that evaluate health care interventions: explanation and elaboration. PLoS Med 2009; 6: e1000100.Google Scholar
42 . Statistical power analysis for the behavioral sciences, 2nd edn. Hillsdale: Lawrence Erlbaum Associates, 1988.Google Scholar
43 . A double-blind comparison of amisulpride vs. haloperidol in acute schizophrenic patients. In: Pichot PBerner PWolf RThau K, eds. Psychiatry, the state of the art. New York: Plenum Press, 1985: 687–91.Crossref, Google Scholar
44 . Switching from clozapine to zotepine in patients with schizophrenia: a 12-week prospective, randomized, rater blind, and parallel study. J Clin Psychopharmacol 2013; 33: 211–14.Crossref, Google Scholar
45 . Diagnosis and drug treatment of psychiatric disorders. Baltimore: Williams and Wilkins, 1969.Google Scholar
46 . Paliperidone for schizophrenia. Am J Health Syst Pharm 2008; 65: 403–13.Crossref, Google Scholar
47 . A meta-analysis of head-to-head comparisons of second-generation antipsychotics in the treatment of schizophrenia. Am J Psychiatry 2009; 166: 152–63.Crossref, Google Scholar
48 . Neuroleptic-induced supersensitivity psychosis: clinical and pharmacologic characteristics. Am J Psychiatry 1980; 137: 16–21.Crossref, Google Scholar
49 . Spontaneous movement disorders in antipsychotic-naive patients with first-episode psychoses: a systematic review. Psychol Med 2009; 39: 1065–76.Crossref, Google Scholar
50 . Amisulpride overdose is frequently associated with QT prolongation and torsades de pointes. J Clin Psychopharmacol 2010; 30: 391–95.Crossref, Google Scholar
51 . Safety of sertindole versus risperidone in schizophrenia: principal results of the sertindole cohort prospective study (SCoP). Acta Psychiatr Scand 2010; 122: 345–55.Crossref, Google Scholar
52 . Comparative mortality associated with ziprasidone and olanzapine in real-world use among 18154 patients with schizophrenia: the ziprasidone observational study of cardiac outcomes (ZODIAC). Am J Psychiatry 2011; 168: 193–201.Crossref, Google Scholar
53 . Antipsychotic-induced hyperprolactinaemia: mechanisms, clinical features and management. Drugs 2004; 64: 229–314.Crossref, Google Scholar
54 . Amisulpride versus flupentixol in schizophrenia with predominantly positive symptomatology—a double-blind controlled study comparing a selective D-2-like antagonist to a mixed D-1-/D- 2-like antagonist. Psychopharmacology (Berl) 1998; 137: 223–32.Crossref, Google Scholar
55 . Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. N Engl J Med 2005; 353: 1209–23.Crossref, Google Scholar
56 . Randomized controlled trial of the effect on quality of life of second- vs first-generation antipsychotic drugs in schizophrenia—cost utility of the latest antipsychotic drugs in schizophrenia study (CUtLASS 1). Arch Gen Psychiatry 2006; 63: 1079–86.Crossref, Google Scholar
57 . Perphenazine for schizophrenia. Cochrane Database Syst Rev 2005; 1: CD003443.Crossref, Google Scholar
58 . Metabolic and cardiovascular adverse effects associated with antipsychotic drugs. Nat Rev Endocrinol 2012; 8: 114–26.Crossref, Google Scholar
59 . Effect sizes in cumulative meta-analyses of mental health randomized trials evolved over time. J Clin Epidemiol 2004; 57: 1124–30.Crossref, Google Scholar
60 . Mid-term and long-term efficacy and effectiveness of antipsychotic medications for schizophrenia: a data-driven, personalized clinical approach. J Clin Psychiatry 2011; 72: 1616–27.Crossref, Google Scholar

