
Bipolar Disorder: From Genes to Behavior Pathways
Abstract
Bipolar disorder (BPD) is a devastating illness that is characterized by recurrent episodes of mania and depression. In addition to these cyclic episodes, individuals with BPD exhibit changes in psychovegetative function, cognitive performance, and general health and well being. In this article we draw from neuroimaging findings in humans, postmortem data, and human genetic and pharmacological studies as well as data from animal models of behavior to discuss the neurobiology of BPD. We conclude with a synthesis of where the field stands and with suggestions and strategies for future areas of study to further increase our conceptual understanding of this complex illness.
(Reprinted with permission from The Journal of Clinical Investigation 2009;119:726–736)
Introduction
Bipolar disorder (BPD), classified as a mood disorder in the Diagnostic and statistical manual of mental disorders (4th edition), is a common, chronic, and recurring medical disorder that is characterized by episodes of mania—extremely elevated mood, energy, unusual thought patterns, and sometimes psychosis—and depression. Although these episodes are usually interspersed with periods of relatively normal mood, BPD is the cause of significant suffering for both patients and their families. BPD leads to limited functioning, which often results in decreased productivity in both the personal and the professional arenas of the patient's life. The prognosis for patients with BPD is poor, with high rates of relapse, lingering residual symptoms, cognitive impairments, and diminished well being (1). Moreover, individuals with BPD frequently have coexisting medical conditions, such as obesity, cardiovascular disease, diabetes mellitus, and thyroid dysfunction, all of which are exacerbated by their BPD symptoms (2).
The prevalence of BPD was thought to be around 1%, but current reported diagnoses indicate that this figure may be closer to 5%. This increased prevalence is mainly accounted for not by an increase in diagnosis of full-blown BPD (which is known as BPD I), but by various softer (i.e., less severe) conditions that fall under the BPD spectrum. Disorders under the BPD spectrum have been grouped based on some overlap in clinical manifestations; however, whether they share the same underlying genetics and pathophysiology is uncertain. Therefore, we focus on the emerging neurobiology surrounding BPD I, referred to herein simply as BPD (3).
Because of the elevated morbidity and mortality suffered by individuals with the disorder, BPD has been increasingly recognized as a major health problem. Despite advances in its diagnosis and recognition, the underlying neurobiology of BPD remains largely unknown. It is thought that BPD is a multifactorial disease that results from a combination of different genetic profiles, characterized by the presence of various protective and/or preventive genes relative to susceptibility and/or risk genes as well as environmental influences, including chronic stressors and traumatic experiences.
Historically, the brain systems receiving the greatest attention in neurobiological studies of BPD have been the monoaminergic neurotransmitter systems, i.e., the serotonergic, noradrenergic, and dopaminergic neurotransmitter systems. These neurotransmitter systems are extensively distributed throughout the brain's network of limbic, striatal, and prefrontal cortical neuronal circuits and are thought to support the behavioral and visceral manifestations of mood disorders (Figure 1). Despite evidence showing that many of these circuits are likely to be involved, no obvious degeneration or complete dysfunction of any single neurotransmitter system has been identified. In this regard, the biological underpinnings of BPD appear to differ from classic neurodegenerative disorders such as Parkinson disease and Alzheimer disease, where clear deficits can be traced to the dopaminergic and cholinergic pathways, respectively. However, as we discuss here, many researchers believe that BPD arises from modulation of synaptic and neural plasticity in critical circuits mediating affective and cognitive function. Thus, BPD may represent a disorder of altered synapses and circuits, rather than being the result of imbalances in individual neurotransmitters.
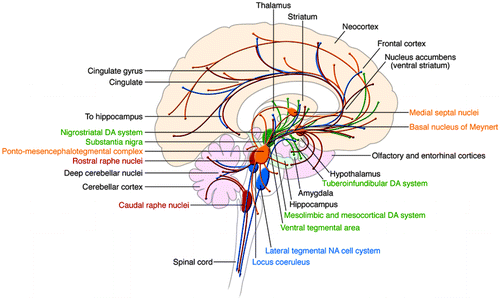
Nuclei as well as their projections are color coded: yellow, cholinergic; green, dopaminergic; blue, noradrenergic; red, serotonergic.
In this Review, we discuss how the currently employed research strategies have furthered our understanding of the underlying neurobiology of BPD. We have broadly categorized these strategies into three main areas: patient-based research including neuroimaging, postmortem tissue analyses, and genetic association studies; analysis of pre-clinical animal models of BPD, such as models of stress and/or depression, models of mania, and, more recently, genetic models that resemble specific facets of BPD symptomatology; and molecular and cellular pharmacologic studies that attempt to identify the cellular and molecular effects of validated moodstabilizing drugs (MSDs). We end with a conclusion about where the field stands, both clinically and pre-clinically, and discuss what we believe is necessary to make the next steps forward. Clearly, we are at the early stages of understanding the neurobiology of this complicated and multi-factorial disease. However, better understanding of the underlying biology is critical for the future development of targeted therapies that are both more effective as well as free from harmful and/or intolerable side effects.
Patient-based research
Neuroimaging.
Functional neuroimaging studies have identified abnormalities in the brains of individuals with BPD, which may be indicative of dysfunction in key neural circuits (4). These circuits are distributed throughout a wide array of neuronal structures, including the amygdala and related limbic nuclei, the orbital and medial prefrontal cortex (PFC), the anterior cingulate, the medial thalamus, and related regions of the basal ganglia (Figure 2) (5). It is plausible that dysfunction or modulation of these circuits may predispose an individual toward manifestation of the symptoms of BPD. For example, studies have shown reduced activity in the right PFC during episodes of mania (6), and dysfunction of the right PFC is thought to contribute to a disinhibited profile, including poor impulse control, risk taking, distractibility, poor sustained attention, and delusions, all of which resemble the symptoms of mania (4, 7).
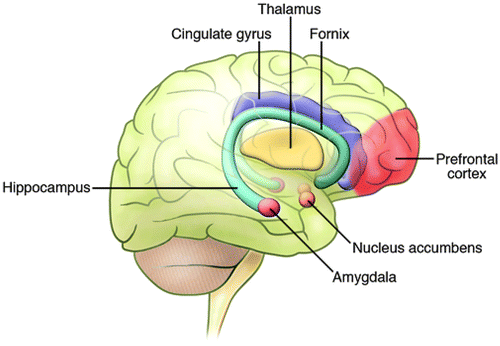
Neuroimaging studies, observations from patients with selective CNS lesions, as well as data from animal behavioral studies have implicated several regions throughout the brain in the control of mood states and emotions. These regions are located throughout the limbic, striatal, and frontal regions. Adapted with permission from Neuropsychopharmacology (24).
Researchers using CT and MRI have identified structural changes in the brains of patients with mood disorders, including patients with BPD. Overall, gray matter volume is not substantially different in patients with BPD compared with normal healthy individuals (8–10). However, several studies have found region-specific reductions in gray matter volume, including increased ventricular size and decreased frontal cortical volume (11, 12). Specifically, volumetric decreases have been identified in the anterior cingulate cortex (13), and other studies have indicated gray matter loss in the left dorsolateral PFC (DL-PFC) (14), the ventral PFC, and the orbital PFC (15). Temporal lobe regions, including the hippocampus and the amygdala, have not been as well studied. However, one study found more prevalent volumetric decreases in the right hippocampal formation of the affected twin in monozygotic twin sets discordant for BPD (16).
White matter hyperintensities (WMHs) around the ventricles and in the subcortical white matter have consistently been found in the brains of depressed elderly persons as well as in the brains of patients with BPD (17, 18). Although the functional and pathological relevance of WMHs has not been fully determined, WMHs have been associated with cerebrovascular accidents (stroke and transient ischemic attacks), ischemia, axonal loss, increased perivascular space, minute brain cysts, and necrosis (19). Recent data suggest that a substantial proportion of individuals with BPD, including children diagnosed with BPD, exhibit WMHs more frequently than the general population. The incidence of WMHs is higher in children with neuropsychiatric disorders, including children with BPD (20, 21). In addition, WMHs are associated with poor treatment outcome in patients with mood disorders, particularly when localized to the subcortical rather than the periventricular areas (22, 23). Combined studies on the increased incidence of WMHs in affective disorders suggest that these lesions could indicate some type of damage to brain tissue, which could result in disruption of the neuronal connections necessary for normal behavioral functioning (24).
Imaging studies using magnetic resonance spectroscopy (MRS) led to the notion that BPD may be associated with mitochondrial dysfunction (25, 26). In particular, high-resolution 1H-MRS imaging studies conducted to quantitatively assess concentrations of N-acetyl-aspartate (NAA), a predominant neurochemical in the human brain that is localized to mature neurons and synthesized within mitochondria, found decreased concentrations of NAA in the hippocampus, the DL-PFC, the orbitofrontal cortex, and the basal ganglia in various patient populations, including patients with BPD (27–32). In addition, studies using 31P-MRS, which allows examination of energy metabolism in the brain, showed a decrease in phosphocreatine and/or ATP levels in patients with a mood disorder, including patients with BPD (33–35). Consistent with these findings, low brain pH levels, as measured indirectly by 31P-MRS, have also been identified in patients with a mood disorder (33–35).
While it is clear that BPD is not a mitochondrial disorder per se, some data suggest that cellular and molecular abnormalities present in the brain of patients with BPD may be associated with alterations in normal mitochondrial function (36). Evidence from microarray, biochemical, neuroimaging, and postmortem studies support the role of mitochondrial dysfunction in BPD (37). In addition to energy production, neuronal mitochondria play an important role in regulating apoptosis, intracellular calcium levels, and synaptic plasticity (24). Mitochondrial dysfunction may also be involved in the calcium signaling abnormality found in BPD (37). Regulation of intracellular calcium levels may be particularly important in the context of the CNS, since fast changes in the levels of calcium are responsible for mediating actions associated with the release of, and response to, neurotransmitters. Changes in calcium levels are also crucial for initiating transcription events at several genes whose transcription is dependent on increases in neuronal activity. One of these neuronal activity-dependent genes, brain-derived neurotrophic factor (BDNF), has been implicated in the etiology of several mood disorders (38). Furthermore, it is known that mitochondria are involved in the initiation of apoptotic processes (39), and newer evidence has suggested that proper mitochondrial function may be important for the regulation of synaptic plasticity. For example, increased neural activity has been shown to induce transcription of genes encoded by mitochondrial DNA, suggesting that increases in energy production may play a role in the regulation of synaptic strength (40).
A role for the glutamatergic system in mood disorders, including BPD, has also been supported by neuroimaging data. In a proton MRS study of children with BPD, patients were shown to have increased levels of glutamate in the frontal lobes and basal ganglia (41), while another study identified increased glutamate levels in the occipital cortex of depressed adult patients (42). Although current data from neuroimaging studies are interesting, their interpretation remains incomplete and often controversial. Despite finding differences in specific locations and deficits in biochemical markers, it is not yet understood what these findings represent or how they may affect the function of various circuits in the brain. In addition, it is not yet known whether the abnormalities that have been discovered represent developmental problems that confer vulnerability to severe mood disorders, compensatory mechanisms for other related pathogenetic processes, or the continual recurrence of affective episodes.
Analysis of postmortem human tissue.
Studies of postmortem brain tissue from patients with recurrent mood disorders, including some with BPD, have found reduced subcortical nuclei volumes (43). Various other studies have shown differential neuronal densities and morphologies that appear to be layer and cell-type specific in both patients with BPD and patients with major depressive disorder. For example, the density of neurons containing large cell somas, which are likely to correspond to glutamatergic pyramidal neurons, was decreased in layers III and V of the DL-PFC of individuals with recurrent mood disorders (44). Furthermore, the size of DL-PFC neurons in layers V and VI was also reduced (45). In patients with BPD, the size of the cell soma of pyramidal neurons in the CA1 region of the hippocampus has been found to be decreased (46), as have cell densities in layers III–VI in different regions of the anterior cingulate (24, 36). Interestingly, cell densities of nonpyramidal neurons were decreased in layer II, and neuron size was increased in layers II and V of these anterior cingulate regions (24, 36).
In addition, decreased levels of calbindin and parvalbumin-expressing neurons, both of which are subtypes of GABAergic interneurons, have been identified in the anterior cingulate cortex, the hippocampus, and the entorhinal cortex of patients with BPD (47, 48). These data, in combination with studies showing decreased hippocampal expression of glutamic acid decarboxylase 67 (GAD67) and somatostatin in the hippocampus of patients with BPD, have led to the hypothesis that a subset of hippocampal interneurons may be abnormal in BPD (49).
In addition to changes in the size and density of neuronal cell types, changes in glial cell biology have also been observed in studies of postmortem tissue from patients with BPD. The density of glial cells seems to be decreased in frontal cortical areas, while the nucleus size increases (44, 50–52). Moreover, a proteomics study using brains from individuals with BPD and individuals with major depressive disorder found disease-specific alterations in levels of glial-fibrillary acidic protein (GFAP), an abundantly expressed astrocyte-specific protein (53). Additional studies have found reductions in oligodendrocyte number and in the expression of genes that are related to oligodendrocyte differentiation and myelin production in the DL-PFC of individuals with BPD (54). The recurrent theme of decreased cell density and number may represent cell loss and atrophy in these patients over the course of disease progression. It is presently unknown whether this type of brain atrophy is one of the underlying causes of the disease, or whether it contributes to illness pathology by disrupting the normal circuitry that is key to normal affective and cognitive functioning.
As discussed above, neuroimaging studies have provided suggestive evidence for abnormalities in mitochondrial function and energy production in BPD. Analysis of expression levels of key mitochondrial-related genes in the postmortem human brain has provided additional evidence to support this idea. A recent microarray study comparing postmortem hippocampal tissue from individuals with BPD and individuals with schizophrenia as well as normal healthy controls revealed that the expression of 43 genes was decreased in patients with BPD compared with those with schizophrenia (49). Furthermore, 42% of these genes encoded mitochondrial proteins.
In addition to the prominent findings of altered expression of genes encoding mitochondrial proteins, substantial changes have been observed in the level of expression of proteins implicated in synaptic function. These findings suggest alterations in synaptic plasticity mechanisms in individuals with BPD. The neuronal plasticity marker GAP-43 is highly expressed in axonal growth cones during development and is implicated in regulation of axonal morphology and synaptic plasticity in the mature brain (55). In patients with BPD, it has been reported that levels of GAP-43 are reduced in both the cingulate cortex and the hippocampus (56, 57). The synapsin family of proteins binds synaptic vesicles to the cytoskeleton, preventing their transport to the presynaptic membrane and subsequent neurotransmitter release (58). Docking of synaptic vesicles and neurotransmitter release are regulated by a complex of proteins that includes SNAP-25, syntaxin, synaptobrevin, and synaptophysin. In postmortem brains from individuals with BPD, a reduction in the levels of synapsin family members has been found in the hippocampus (59). However, increases in the levels of SNARE complex proteins, which are responsible for mediating the fusion of synaptic vesicles with the cell membrane, have been observed in the DL-PFC (60). An additional study showed decreased levels of synaptobrevin and synaptophysin in the visual association cortex of patients with BPD (61). The levels of mRNAs encoding netrins, a family of proteins important in regulating axon guidance, have also been shown to be reduced in the CA3 region of the hippocampus and entorhinal cortex of patients with BPD (62).
Genetic studies.
Combined evidence from family, twin, and adoption studies suggests that BPD has a strong genetic component. Twin studies show a highly elevated concordance rate in monozygotic twins when compared with dizygotic twins (63, 64), and BPD is more likely to occur in biological parents of adopted children than in the adoptive parents (65). Strategies for detecting the genetics of BPD include both linkage and association studies. Linkage methods test the loci of vulnerability genes by studying co-inheritance of chromosomal fragments in specific illnesses, whereas association studies test whether a specific gene variant is associated with a given disorder. A list of genes, including their known functions and potential role in the etiology of BPD, that have been implicated via both linkage and association studies is lengthy and beyond the scope of this review (see ref 66 for an indepth discussion). Of the implicated genes, most attention has been given to those that encode proteins known to interact with signaling pathways previously implicated in BPD. For example, several of the risk genes that have been identified are known to interact with the PKC and glycogen synthase kinase 3β (GSK3β) signaling pathways (67, 68). In addition, other putative susceptibility genes, including glutamate receptor, metabotropic 3 (GRM3) and GRM4; glutamate receptor, ionotropic, N-methyl-D-aspartate 2B (GRIN2B); D-amino acid oxidase (DAO); and DAO activator (DAOA, also known as G72), encode proteins involved in glutamatergic signaling (68, 69). In addition, a number of genes encoding proteins involved in circadian biology, including ARNT-like protein 1, brain and muscle (BmaL1), TIMELESS, and PERIOD3, have been implicated as susceptibility genes for BPD (68). These genes are of interest because virtually all patients with BPD have alterations in circadian function, including alterations in sleep patterns, activity, hormonal secretions, and appetite.
Beyond the older linkage and association studies, recent advances in technology have allowed researchers to study the genetic component of BPD using genome-wide association studies (GWASs). Four groups have recently performed independent GWASs of BPD (67, 70–72). However, the significance of these findings is unclear, since very few findings have been replicated from sample to sample and there is the possibility of multiple false positives due to the substantial number of comparisons. In one study using 1,233 patients with BPD and 1,439 control subjects, Baum and colleagues identified a SNP in diacylglycerol kinase η (DGKH) as being associated with BPD (67). The Wellcome Trust Case Control Consortium (WTCC) analyzed 1,868 individuals with BPD and 2,938 control subjects and identified a locus in a gene rich region of high linkage disequilibrium on chromosome 16p12 as being associated with BPD (70). Sklar et al.'s study of 1,461 patients with BPD and 2,008 control subjects found their strongest result in myosin 5B (MYO5B) (71). The most recent GWAS, by Ferreira et al., used a new sample with 1,098 individuals with BPD and 1,267 control subjects and found the strongest results in ankyrin G (ANK3) and calcium channel, voltage-dependent, L-type, α 1C subunit (CACNA1C) (72). A broad comparison of the WTCC and the Sklar study also confirmed the CACNA1C results, identifying CACNA1C as showing a consistently strong signal in individuals with BPD (72). More studies are clearly necessary before any consensus can be made as to the significance of these recent findings.
Analysis of pre-clinical animal models of bpd
While considerable caution undoubtedly needs to be taken in applying animal models to complex neuropsychiatric disorders, animal models can be valuable tools for exploring the underlying pathologies of human diseases and developing better therapies. An ideal animal model for BPD should show face validity, i.e., it should include spontaneous and progressive behavior that oscillates between increased and decreased manifestations of the behavior being modeled, which should be similar to a behavior characteristic of either human mania or depression. Moreover, the modeled behavior should show predictive validity, i.e., it can be normalized by treatment with MSDs (36). The progressive and cyclic nature of BPD presents a unique challenge for modeling in rodents. Indeed, as discussed in detail below, most models have tended to focus on either mania or depression, rather than modeling both behaviors, which both occur in individuals with BPD (36, 73). Currently, more models of depressive illness exist than of mania or of the cyclical nature of BPD (73). Increasing the difficulty of developing animal models of BPD is the hypothesis that an interaction between both genes and the environment are needed in order to manifest symptoms of the illness. Determining what types of environmental disturbances to pair with the various genetic alterations is difficult, but may be critical in effecting useful models.
Models of stress and/or depression.
A commonly used line of research for those studying BPD using animal models is to examine the relationship between cellular and molecular characteristics of animals and behavior in laboratory situations that replicate environments thought to predispose humans toward depression. Some of the more commonly used models have been maternal separation and various chronic stress paradigms including psychosocial stress, mild unpredicted stress, and restraint/immobilization stress (36, 74). Another model used to induce a depression-like state is the learned helplessness model (73, 75). In this model, animals are administered an inescapable aversive stimulus, frequently an electric foot shock. Following the inescapable portion of the paradigm, animals are again administered the aversive stimulus, but in an environment where they are capable of escaping the aversive stimulus. Longer latencies or a lack of any response is seen as indicative of “learned helplessness” (73, 75). The forced swim test is the most commonly used animal model in depression research, and more specifically has been widely used as a screen for antidepressant treatments (74, 76). These models have been useful thus far in helping to screen for potential therapeutic candidates. Further, studies with genetically modified animals using these paradigms have helped to identify candidate molecular and genetic signaling pathways that may be involved in the development of behavioral attributes that mirror disease symptomatology. However, we have yet to be able to adequately model the diseases well enough to determine anything conclusive about the overall etiology of the disorder.
Models of mania.
Hyperactivity is one of the more simple behaviors to detect and quantify in animals. Thus, many models of mania that have been developed have focused on this aspect as the core of their modeling (36, 77). Acute treatment with psychostimulants such as methamphetamines and cocaine can produce a wide range of mania-like behaviors, including hyperactivity, heightened awareness and alertness, insomnia, and changes in sleep patterns (78, 79). Psychostimulant-induced hyperactivity is sensitive to lithium treatment and possibly to other anticonvulsants that have been used as MSDs. Sleep deprivation has also been used with some success as a model for mania. Rats exposed to 72 hours of sleep deprivation exhibit a variety of behaviors that show face validity for modeling human mania. These behaviors include insomnia, hyperactivty, irritability, aggressive behavior, and hypersexuality (78, 79). Furthermore, in terms of predictive validity, lithium has been shown to alleviate some of these mania-like behaviors.
Genetic models of BPD.
Numerous genetic modifications, most of which have been engineered in the mouse species (Table 1), show increased depressive-like symptoms and alterations in affective behavior in various behavioral tests. As previously discussed, depressive-like behavior is often measured as the extent of behavioral desperation in the forced swim test or as increased susceptibility toward learned helplessness. In addition, genetic animal models have been developed that do not confer susceptibility to depression per se but do render the animals unresponsive to antidepressants (Table 2). These models have been useful in helping to identify various genes and signaling pathways that may be involved in the development of depressive symptoms and, importantly, in the response to antidepressant drugs. Here we highlight several findings that have produced much interest and stimulated continued research in the field.
 |
Table 1. Important Genetically Engineered Mice That Model BPD Behaviors.
 |
Table 2. Genetically Engineered Mice Important to BPD Research.
The neurotrophic hypothesis of depression posits that growth factors, in particular BDNF, play a key role in regulating mood (38). This hypothesis arose from several observations: first, that the amount of BDNF is decreased in the hippocampus of animals showing abnormalities in depressive and anxiety-like behavior (80); second, that chronic but not acute antidepressant usage increases BDNF levels (81); and third, that acute application of BDNF, via intrahippocampal injection, has an antidepressant effect (82). However, the neurotrophic hypothesis of depression has been complicated by various genetic manipulations within the BDNF signaling pathway, which show complex, region-specific effects. For example, it has now been shown that contrary to its antidepressant effect in the hippocampus, administration of BDNF in the ventral tegmental area (VTA) results in pro-depressive behaviors (83). Conditional deletion of BDNF in the forebrain does not seem to confer depression per se but does seem to be important in the ability to respond to antidepressants (84). Interestingly, the inability to respond to antidepressants has also been observed in mice that lack adult hippocampal neurogenesis (85). A number of investigators have produced results suggesting that creation of new neurons in the dentate gyrus of the hippocampus is involved in mood regulation (86). Since BDNF has been implicated in regulation of complex mood-related behavior as well as the proliferation and survival of new neurons in the dentate, it has been postulated that BDNF signaling may be an important component in adult neurogenesis and that this pathway may be uniquely involved in the regulation of mood-related behavior (86). However, it is clear that the story is not completely straightforward, and further research is necessary to elucidate the effects of BDNF in the etiology of depression, the role of BDNF in the response to antidepressant treatment, and how these effects may be related to adult hippocampal neurogenesis.
Two recent reports of mutant mice resembling behavioral facets of mania have reinvigorated interest in animal models of BPD (Table 1). In the first report, McClung and colleagues found that mutant mice carrying a Clock gene mutation that encodes a dominant-negative CLOCK protein are hyperactive, need less sleep, and show an increased propensity for stimulants and reward (87). These mutant mice exhibit less depressive and anxiety-like behavior and, remarkably, chronic administration of lithium reduced many of the behavioral features reminiscent of mania. These results are particularly interesting since circadian rhythms and the genes that underlie the molecular clock have been highly implicated in BPD and because patients with BPD show marked behavioral impairments in their circadian rhythms. A second report of interest concerns the gene encoding glutamate receptor 6 (Glur6) (101). As GLUR6 resides in a genetic linkage region (6q21) associated with BPD, the demonstration that Glur6-deficient mice exhibit behavior related to symptoms of mania is very exciting (88). Specifically, Glur6-deficient mice exhibit less anxious behavior, more risk-taking behavior, less despair-type manifestations, and increased aggressive displays. Further, chronic lithium treatment in these mice reduces hyperactivity, aggression, and some risk-taking behavior (88). Future studies similar to these should begin to shed light on the etiology of BPD as well as provide various genetic and behavioral models that can be used to dissect the underlying neurobiological mechanisms associated with particular deficits in BPD.
Recent studies have identified a molecule that may be associated with affective resilience and enhanced recovery from depressive-like and manic-like states (89). The molecule, Bcl-2-associated athanogene (Bag1), interacts with Hsp70, glucocorticoid receptors (GRs), Bcl-2, and Raf, thereby regulating intracellular signal transduction, nuclear hormone receptors, gene transcription, and cell survival. Microarray studies identified Bag1 as a target for the actions of MSDs (90), and analysis of neuron-selective Bag1 transgenic mice indicated a beneficial role for the protein in depressive-like and manic-like behaviors (102). Specifically, Bag1 transgenic mice showed less anxious-like behavior and had higher spontaneous recovery rates following analysis in the learned helplessness model of stress and/or depression. On mania-related tests, Bag1 transgenic mice recovered much faster in the amphetamineinduced hyperlocomotion test and displayed a clear resistance to cocaine-induced behavioral sensitization.
Molecular and cellular pharmacologic studies
Despite substantial advances in pharmacotherapeutics, lithium remains the gold standard therapy for BPD. Valproic acid (VPA), which was first used therapeutically as an anticonvulsive, has become an additional mainstay for treating BPD (36). Current research on understanding the actions of lithium and VPA assumes that these drugs do not have major direct interactions with cell surface receptors and are more likely to exert their effects directly or indirectly through modulation of intracellular targets. Although several targets and mechanisms of these drugs have been identified, precisely which effects are responsible for specific facets of the therapeutic response remains to be fully elucidated. Since the chemical structures of lithium, which is a monovalent cation, and VPA, which is a fatty acid, are highly dissimilar, identifying common targets for these two effective MSDs could be potentially important, as shared biochemical targets would be more likely to represent targets underlying their therapeutic value. It is beyond the scope of this article to review all the molecular and cellular pharmacologic studies; the reader is referred to other reviews (91, 92). Here, we evaluate the most extensively studied and highly prominent targets.
Lithium at therapeutically relevant concentrations functions as a direct inhibitor of a limited number of enzymes because it acts as a competitor for magnesium (93, 94). The enzymes known to be inhibited by lithium include inositol monophosphatase (IMPase); inositol polyphosphate 1-phosphatase (IPPase); bisphosphate 3′-nucleotidase; fructose 1,6-bisphosphatase; GSK3β; and phosphoglucomutase (reviewed in refs. 95–97). The signaling pathways that involve IMPase and GSK3, the phosphoinositol signaling cascade and the Wnt signaling pathway, respectively, have received the greatest attention and interest (Figure 3).
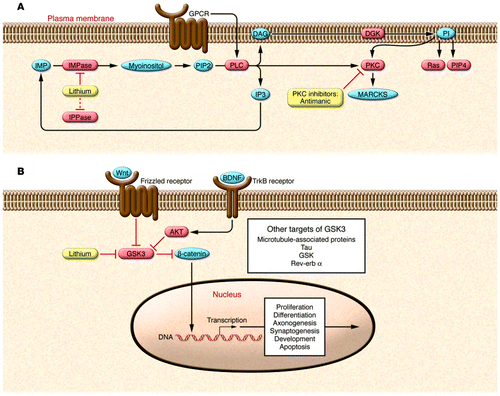
(A) Lithium directly inhibits key enzymes including IPPase and IMPase that regulate inositol-1,4,5-triphosphate (IP3) recycling. IMPase-mediated catalysis represents the final step in the conversion of inositol monophosphate (IMP) to myoinositol. Thus, inhibiting IMPase can reduce myoinositol levels. The inositol depletion hypothesis proposes that IMPase inhibition interferes with PI synthesis. The PI signaling cascade starts with surface receptor activation, shown here as GPCR-mediated activation of PLC. Activated PLC catalyzes the hydrolysis of PI biphosphate (PIP2) to diacylglycerol (DAG) and IP3. DAG activates PKC, which, among many other functions, activates myristoylated, alanine-rich C-kinase substrate (MARCKS). The antimanic drugs lithium and VPA both decrease levels of phosphorylated and total MARCKS. Molecules in red depict enzymes, while molecules in blue depict second messengers. PI signaling modulates other second messenger proteins including the small GTPase, Ras, and phosphatidylinositol-4-phosphate (PIP4). (B) Overview of the Wnt and GSK3 signaling pathways. Both lithium and Wnt signaling can inhibit GSK3. In the Wnt signaling pathway, Wnt glycoproteins interact with the frizzled family of receptors to stimulate the dishevelled-mediated (not shown) inactivation of GSK3. Inhibition of GSK3 prevents β-catenin phosphorylation, which inhibits its degradation and allows it to act as a transcription factor. Wnt proteins have been implicated in the regulation of neuron morphology, neurotransmission, and synaptogenesis. GSK3 is also inhibited by AKT (also known as PKB), which is activated downstream of the tropomyosin receptor B (TrkB), whose ligand is BDNF. In addition to inhibiting β-catenin, GSK3 has numerous other targets including microtubule-associated proteins, tau, GSK, and rev-erb α. Adapted with permission from Neuropsychopharmacology (24).
The phosphoinositol signaling cascade mediated by G protein activation of PLC-B is one of the most extensively studied pathways in BPD research (Figure 3A). The direct inhibition of IMPase and IPPase by lithium suggests that it may decrease the bioavailability of myoinositol. This inositol depletion hypothesis posits that lithium interferes with the regeneration of inositol and, under conditions where inositol limits phosphatidylinositol (PI) synthesis, depletes the cell of PI. Because PI is an obligate precursor of PI biphosphate (PIP2), the hypothesis suggests that inhibition of IMPase could disrupt PIP2/IP3-mediated signaling (98, 99). Although the inositol depletion hypothesis provides an elegant potential mechanism to explain the actions of lithium, several important issues remain unresolved, and recent data have hallenged some of the predilections to the model. For example, therapeutic levels of lithium do not deplete PIP2 in vivo, and this phenomenon is an essential component of the hypothesis (100). Moreover, although the in vivo inhibition of IMPase is incomplete, it is not clear whether this modest reduction in inositol is sufficient to impair PI or PIP2 synthesis (101). Research has therefore moved to more downstream targets in the PI signaling cascade. It has been demonstrated that lithium reduces the levels of specific PKC isozymes in limbic and limbic-related areas of the brain. Notably, VPA also exerts similar attenuating effects on PKC signaling, whereas agents capable of triggering mania exert the opposite effects. One of the genes emerging as a risk factor for BPD in GWASs, DGKH, encodes a protein that is an upstream regulator of PKC signaling. These findings on the PKC pathway have led to the use of tamoxifen, as high concentrations directly inhibit PKC, with promising preliminary results in the treatment of mania (102). These data suggest that direct, centrally acting PKC inhibitors may represent novel therapeutics for the treatment of mania (92, 103).
GSK3 antagonizes both the insulin and Wnt signaling pathways and has recently been demonstrated to regulate synaptic plasticity (104–112). Many of the known effects of lithium can theoretically be explained in terms of GSK3 inhibition. For example, lithium increases neuronal growth cone area (104, 105), alters synaptogenesis (106, 107), and stimulates hippocampal neurogenesis (108). All of these effects mimic Wnt signaling (107, 109, 110). However, inhibition of GSK3 is achieved at the higher end of the therapeutic range of lithium, raising some questions about whether this level of inhibition is sufficient to have strong biological effects. However, secondary inhibition modes have been proposed, which may enhance direct inhibition of GSK3 by lithium (111, 112). Pharmacologic and genetic approaches that regulate GSK3 have behavioral effects that suggest utility in the treatment of BPD. For example, two structurally dissimilar GSK3 small molecule inhibitors and a peptide inhibitor reduce immobility in the forced swim test and attenuate amphetamine-induced hyperactivity (113, 114).
In addition to its direct effects on GSK3, chronic lithium administration at therapeutically relevant concentrations induces prominent neuroprotective and neurotrophic proteins, including Bcl-2 (115) and BDNF (116), in rodents and cultured neurons. Bcl-2 is not only a major antiapoptotic protein, but also stimulates axonal regeneration following injury (reviewed in ref. 117). Consistent with its effects on GSK3, BDNF, and Bcl-2, lithium has been dem- onstrated to exert robust neuroprotective properties against various insults both in vitro and in vivo (reviewed in ref. 118). Notably, several independent studies demonstrated that lithium has neuroprotective effects in animal and cellular models of Alzheimer disease, Huntington disease, Parkinson disease, retinal degeneration, spinal cord injury, and HIV infection (119). Finally, human neuroimaging data have shown that chronic lithium treatment in patients with BPD increases NAA levels (a putative marker of neuronal viability) as well as regional gray matter volumes (120, 121). These human data suggest that activation of neurotrophic cascades may underlie the mechanisms by which this simple monovalent cation exerts major effects in the treatment of BPD.
VPA has clear effects on high-frequency sodium channel firing and GABA function, effects that are likely to contribute to its effects as an anticonvulsant and potentially to its effects as a mood stabilizer (91). Its ability to function as a histone deacetylase (HDAC) inhibitor has received a great deal of attention, as the role of epigenetic modification and transcription regulation in the control of mood behaviors has garnered increased visibility (122). Recent research has fostered the idea that epigenetic mechanisms, exerting long-lasting control over gene expression, could mediate stable changes in brain function in individuals with BPD. Acetylation of histone tail lysines is a major epigenetic mechanism that is generally associated with transcriptional activation. Recently, Weaver and colleagues showed that the epigenomic state of the GR gene could be established via behavioral programming in rats (123). Remarkably, this epigenomic state could be reversed with an HDAC inhibitor (124). Thus, it has been postulated that earlylife environmental stressors contributing to the development of or predisposition to BPD, might be responsive to HDAC inhibitors.
Conclusions and strategies for the future
The large body of literature about the development and underlying neurobiology of BPD indicates a potential role for a multitude of different signaling pathways and neuroendocrine systems. As we have reviewed here, there is an emerging body of evidence suggesting that abnormalities in the regulation of signaling and neural plasticity are integral to the underlying neuropathology of BPD. A correction of dysregulated trans-synaptic signaling by MSDs represents a physiological process that curtails the major oscillations in behavioral states associated with BPD. However, determining which abnormalities actually mediate predisposition to the disease and which are merely factors that are along for the ride seems difficult with so many different pathways and genes being implicated. Although recent research has made great strides toward a better understanding of the illness, continued technological progression in genetic studies and animal models of behavior hold promise for extending our knowledge in the future.
An important question to address in future research concerns the distinction between acute, subchronic, and chronic effects of MSDs and the environmental stressors that serve to predispose to or contribute to illness development. A clear understanding of the timeline under which stressors and MSDs mediate their effects will help guide future studies, enabling researchers to develop hypotheses to test the mechanisms underlying known effects. For example, acute and subchronic effects are most likely to be mediated by regulation of neurotransmission, kinase activity, and enzyme inhibition or facilitation. On the other hand, chronic effects are most likely to be mediated by more stable, long-lasting changes rooted in changes in gene transcription, likely as a result of epigenetics and chromatin remodeling or changes to signaling loops as a result of continual feedback pressure.
As discussed above, a wealth of neuroimaging, neuropathological, biochemical, and behavioral studies over the course of the past decade have suggested numerous candidates for impairments in BPD, including a preponderance of components of various cellular plasticity cascades. These results highlight the potential importance of neuroprotective and neurotrophic effects in the etiology and treatment of BPD. A growing list of genes that confer susceptibility to BPD have been identified, a number of which have been implicated in the cellular or molecular pathophysiology of the disease. These data and the promise of future technological advances hold much promise for the potential discovery of novel therapeutics for BPD.
1. Belmaker, R.H. 2004. Bipolar disorder. N. Engl. J. Med. 351:476–486.Crossref, Google Scholar
2. Kupfer, D.J. 2005. The increasing medical burden in bipolar disorder. JAMA. 293:2528–2530.Crossref, Google Scholar
3. Akiskal, H.S., et al. 2000. Re-evaluating the prevalence of and diagnostic composition within the broad clinical spectrum of bipolar disorders. J. Affect. Disord. 59(Suppl. 1):S5–S30.Crossref, Google Scholar
4. Wilkins, A.J., Shallice, T., and McCarthy, R. 1987. Frontal lesions and sustained attention. Neuropsychologia. 25:359–365.Crossref, Google Scholar
5. Carlson, P.J., Singh, J.B., Zarate, C.A., Drevets, W.C., and Manji, H.K. 2006. Neural circuitry and neuroplasticity in mood disorders: insights for novel therapeutic targets. NeuroRx. 3:22–41.Crossref, Google Scholar
6. Blumberg, H.P., et al. 1999. Rostral and orbital prefrontal cortex dysfunction in the manic state of bipolar disorder. Am. J. Psychiatry. 156:1986–1988.Google Scholar
7. Woods, D.L., and Knight, R.T. 1986. Electrophysiologic evidence of increased distractibility after dorsolateral prefrontal lesions. Neurology. 36:212–216.Crossref, Google Scholar
8. Brambilla, P., et al. 2001. Differential effects of age on brain gray matter in bipolar patients and healthy individuals. Neuropsychobiology. 43:242–247.Crossref, Google Scholar
9. Schlaepfer, T.E., et al. 1994. Decreased regional cortical gray matter volume in schizophrenia. Am. J. Psychiatry. 151:842–848.Crossref, Google Scholar
10. Zipursky, R.B., et al. 1997. Deficits in gray matter volume are present in schizophrenia but not bipolar disorder. Schizophr. Res. 26:85–92.Crossref, Google Scholar
11. Nasrallah, H.A., McCalley-Whitters, M., and Jacoby, C.G. 1982. Cortical atrophy in schizophrenia and mania: a comparative CT study. J. Clin. Psychiatry. 43:439–441.Google Scholar
12. Pearlson, G.D., and Veroff, A.E. 1981. Computerised tomographic scan changes in manic-depressive illness. Lancet. 2:470.Crossref, Google Scholar
13. Drevets, W.C., et al. 1997. Subgenual prefrontal cortex abnormalities in mood disorders. Nature. 386:824–827.Crossref, Google Scholar
14. Brambilla, P., et al. 2002. Anatomical MRI study of subgenual prefrontal cortex in bipolar and unipolar subjects. Neuropsychopharmacology. 27:792–799.Crossref, Google Scholar
15. Frangou, S., Raymont, V., and Bettany, D. 2002. The Maudsley bipolar disorder project. A survey of psychotropic prescribing patterns in bipolar I disorder. Bipolar Disord. 4:378–385.Crossref, Google Scholar
16. Noga, J.T., Vladar, K., and Torrey, E.F. 2001. A volumetric magnetic resonance imaging study of monozygotic twins discordant for bipolar disorder. Psychiatry Res. 106:25–34.Crossref, Google Scholar
17. Altshuler, L.L., et al. 1995. T2 hyperintensities in bipolar disorder: magnetic resonance imaging comparison and literature meta-analysis. Am. J. Psychiatry. 152:1139–1144.Crossref, Google Scholar
18. Stoll, A.L., Renshaw, P.F., Yurgelen-Todd, D.A., and Cohen, B.M. 2000. Neuroimaging in bipolar disorder: what have we learned? Biol. Psychiatry. 48:505–517.Crossref, Google Scholar
19. Ovbiagele, B., and Saver, J.L. 2006. Cerebral white matter hyperintensities on MRI: Current concepts and therapeutic implications. Cerebrovasc. Dis. 22:83–90.Crossref, Google Scholar
20. Lyoo, I.K., Lee, H.K., Jung, J.H., Noam, G.G., and Renshaw, P.F. 2002. White matter hyperintensities on magnetic resonance imaging of the brain in children with psychiatric disorders. Compr. Psychiatry. 43:361–368.Crossref, Google Scholar
21. Pillai, J.J., et al. 2002. Increased presence of white matter hyperintensities in adolescent patients with bipolar disorder. Psychiatry. Res. 114:51–56.Crossref, Google Scholar
22. Lenox, R.H., Gould, T.D., and Manji, H.K. 2002. Endophenotypes in bipolar disorder. Am. J. Med. Genet. 114:391–406.Crossref, Google Scholar
23. Moore, P.B., et al. 2001. Cerebral white matter lesions in bipolar affective disorder: relationship to outcome. Br. J. Psychiatry. 178:172–176.Crossref, Google Scholar
24. Schloesser, R.J., Huang, J., Klein, P.S., and Manji, H.K. 2008. Cellular plasticity cascades in the pathophysiology and treatment of bipolar disorder. Neuropsychopharmacology. 33:110–133.Crossref, Google Scholar
25. Hamakawa, H., et al. 2004. Reduced intracellular pH in the basal ganglia and whole brain measured by 31P-MRS in bipolar disorder. Psychiatry Clin. Neurosci. 58:82–88.Crossref, Google Scholar
26. Kato, T., et al. 1998. Decreased brain intracellular pH measured by 31P-MRS in bipolar disorder: a confirmation in drug-free patients and correlation with white matter hyperintensity. Eur. Arch. Psychiatry Clin. Neurosci. 248:301–306.Crossref, Google Scholar
27. Hamakawa, H., Kato, T., Murashita, J., and Kato, N. 1998. Quantitative proton magnetic resonance spectroscopy of the basal ganglia in patients with affective disorders. Eur. Arch. Psychiatry Clin. Neurosci. 248:53–58.Crossref, Google Scholar
28. Bertolino, A., et al. 2003. Neuronal pathology in the hippocampal area of patients with bipolar disorder: a study with proton magnetic resonance spectroscopic imaging. Biol. Psychiatry. 53:906–913.Crossref, Google Scholar
29. Cecil, K.M., DelBello, M.P., Morey, R., and Strakowski, S.M. 2002. Frontal lobe differences in bipolar disorder as determined by proton MR spectroscopy. Bipolar Disord. 4:357–365.Crossref, Google Scholar
30. Chang, K., et al. 2003. Decreased N-Acetylaspartate in children with familial bipolar disorder. Biol. Psychiatry. 53:1059–1065.Crossref, Google Scholar
31. Diecken, R.F., Pegues, M.P., Anzalone, S., Feiwell, R., and Soher, B. 2003. Lower concentration of hippocampal N-acetylaspartate in familial bipolar I disorder. Am. J. Psychiatry. 160:873–882.Crossref, Google Scholar
32. Winsberg, M.E., et al. 2000. Decreased dorsolateral prefrontal N-acetyl aspartate in bipolar disorder. Biol. Psychiatry. 47:475–481.Crossref, Google Scholar
33. Diecken, R.F., Fein, G., and Weiner, M.W. 1995. Abnormal frontal lobe phosphorous metabolism in bipolar disorder. Am. J. Psychiatry. 152:915–918.Crossref, Google Scholar
34. Kato, T., et al. 1995. Lateralized abnormality of high energy phosphate metabolism in the frontal lobes of patients with bipolar disorder detected by phaseencoded 31P-MRS. Psychol. Med. 25:557–566.Crossref, Google Scholar
35. Volz, H.P., et al. 1998. 31P magnetic resonance spectroscopy in the frontal lobe of major depressed patients. Eur. Arch. Psychiatry Clin. Neurosci. 248:289–295.Crossref, Google Scholar
36. Goodwin, F.K., and Jamison, K.R. 2007. Manicdepressive illness: bipolar disorders and recurrent depression. Oxford University Press. New York, New York, USA. 1288 pp.Google Scholar
37. Quiroz, J.A., Gray, N.A., Kato, T., and Manji, H.K. 2008. Mitochondrially mediated plasticity in the pathophysiology and treatment of bipolar disorder. Neuropsychopharmacology. 33:2551–2565.Crossref, Google Scholar
38. Duman, R.S., and Monteggia, L.M. 2006. A neurotrophic model for stress-related mood disorders. Biol. Psychiatry. 59:1116–1127.Crossref, Google Scholar
39. Mattson, M.P. 2007. Mitochondrial regulation of neuronal plasticity. Neurochem. Res. 32:707–715.Crossref, Google Scholar
40. Williams, J.M., Thompson, V.L., Mason-Parker, S.E., Abraham, W.C., and Tate, W.P. 1998. Synaptic activity-dependent modulation of mitochondrial gene expression in the rat hippocampus. Brain Res. Mol. Brain Res. 60:50–56.Crossref, Google Scholar
41. Castillo, M., Kwock, L., Courvoisie, H., and Hooper, S.R. 2000. Proton MR spectroscopy in children with bipolar affective disorder: preliminary observations. AJNR Am. J. Neuroradiol. 21:832–838.Google Scholar
42. Sanacora, G., et al. 2004. Subtype-specific alterations of gamma-aminobutyric acid and glutamate in patients with major depression. Arch. Gen. Psychiatry. 61:705–713.Crossref, Google Scholar
43. Bielau, H., et al. 2005. Volume deficits of subcortical nuclei in mood disorders A postmortem study. Eur. Arch. Psychiatry Clin. Neurosci. 255:401–412.Crossref, Google Scholar
44. Rajkowska, G. 2002. Cell pathology in bipolar disorder. Bipolar Disord. 4:105–116.Crossref, Google Scholar
45. Cotter, D., et al. 2002. Reduced neuronal size and glial cell density in area 9 of the dorsolateral prefrontal cortex in subjects with major depressive disorder. Cerebral Cortex. 12:386–394.Crossref, Google Scholar
46. Liu, L., Schulz, S.C., Lee, S., Reutiman, T.J., and Fatemi, S.H. 2007. Hippocampal CA1 pyramidal cell size is reduced in bipolar disorder. Cell. Mol. Neurobiol. 27:351–358.Crossref, Google Scholar
47. Cotter, D., et al. 2002. The density and spatial distribution of GABAergic neurons, labelled using calcium binding proteins, in the anterior cingulate cortex in major depressive disorder, bipolar disorder, and schizophrenia. Biol. Psychiatry. 51:377–386.Crossref, Google Scholar
48. Pantazopoulos, H., Lange, N., Baldessarini, R.J., and Berretta, S. 2007. Parvalbumin neurons in the entorhinal cortex of subjects diagnosed with bipolar disorder or schizophrenia. Biol. Psychiatry. 61:640–652.Crossref, Google Scholar
49. Konradi, C., et al. 2004. Molecular evidence for mitochondrial dysfunction in bipolar disorder. Arch. Gen. Psychiatry. 61:300–308.Crossref, Google Scholar
50. Ongur, D., Drevets, W.C., and Price, J.L. 1998. Glial reduction in the subgenual prefrontal cortex in mood disorders. Proc. Natl. Acad. Sci. U. S. A. 95:13290–13295.Crossref, Google Scholar
51. Rajkowska, G. 2000. Postmortem studies in mood disorders indicate altered numbers of neurons and glial cells. Biol. Psychiatry. 48:766–777.Crossref, Google Scholar
52. Rajkowska, G., Halaris, A., and Selemon, L.D. 2001. Reductions in neuronal and glial density characterize the dorsolateral prefrontal cortex in bipolar disorder. Biol. Psychiatry. 49:741–752.Crossref, Google Scholar
53. Johnston-Wilson, N.L., et al. 2000. Disease-specific alterations in frontal cortex brain proteins in schizophrenia, bipolar disorder, and major depressive disorder. The Stanley Neuropathology Consortium. Mol. Psychiatry. 5:142–149.Crossref, Google Scholar
54. Uranova, N.A., Vostrikov, V.M., Orlovskaya, D.D., and Rachmanova, V.I. 2004. Oligodendroglial density in the prefrontal cortex in schizophrenia and mood disorders: a study from the Stanley Neuropathology Consortium. Schizophr. Res. 67:269–275.Crossref, Google Scholar
55. Strittmatter, S.M. 1992. GAP-43 as a modulator of G protein transduction in the growth cone. Perspect. Dev. Neurobiol. 1:13–19.Google Scholar
56. Eastwood, S.L., and Harrison, P.J. 2001. Synaptic pathology in the anterior cingulate cortex in schizophrenia and mood disorders. A review and a Western blot study of synaptophysin, GAP-43 and the complexins. Brain Res. Bull. 55:569–578.Crossref, Google Scholar
57. Tian, S.Y., Wang, J.F., Bezchlibnyk, Y.B., and Young, L.T. 2007. Immunoreactivity of 43 kDa growthassociated protein is decreased in post mortem hippocampus of bipolar disorder and schizophrenia. Neurosci. Lett. 411:123–127.Crossref, Google Scholar
58. Hilfiker, S., et al. 1999. Synapsins as regulators of neurotransmitter release. Philos. Trans. R. Soc. Lond. B Biol. Sci. 354:269–279.Crossref, Google Scholar
59. Vawter, M.P., et al. 2002. Reduction of synapsin in the hippocampus of patients with bipolar disorder and schizophrenia. Mol. Psychiatry. 7:571–578.Crossref, Google Scholar
60. Scarr, E., Gray, L., Keriakous, D., Robinson, P.J., and Dean, B. 2006. Increased levels of SNAP-25 and synaptophysin in the dorsolateral prefrontal cortex in bipolar I disorder. Bipolar Disord. 8:133–143.Crossref, Google Scholar
61. Beasley, C.L., et al. 2005. Reductions in cholesterol and synaptic markers in association cortex in mood disorders. Bipolar Disord. 7:449–455.Crossref, Google Scholar
62. Eastwood, S.L., and Harrison, P.J. 2007. Decreased mRNA expression of netrin-G1 and netrin-G2 in the temporal lobe in schizophrenia and bipolar disorder. Neuropsychopharmacology. 33:933–945.Crossref, Google Scholar
63. Bertelsen, A., Harvald, B., and Hauge, M. 1977. A Danish twin study of manic-depressive disorders. Br. J. Psychiatry. 130:330–351.Crossref, Google Scholar
64. Cardno, A.G., et al. 1999. Heritability estimates for psychotic disorders: the Maudsley twin psychosis series. Arch. Gen. Psychiatry. 56:162–168.Crossref, Google Scholar
65. Mendlewicz, J., and Rainer, J.D. 1977. Adoption study supporting genetic transmission in manicdepressive illness. Nature. 268:327–329.Crossref, Google Scholar
66. Hayden, E.P., and Nurnberger, J.I., Jr. 2006. Molecular genetics of bipolar disorder. Genes Brain Behav. 5:85–95.Crossref, Google Scholar
67. Baum, A.E., et al. 2008. A genome-wide association study implicates diacylglycerol kinase eta (DGKH) and several other genes in the etiology of bipolar disorder. Mol. Psychiatry. 13:197–207.Crossref, Google Scholar
68. Newberg, A.R., Catapano, L.A., Zarate, C.A., and Manji, H.K. 2008. Neurobiology of bipolar disorder. Expert Rev. Neurother. 8:93–110.Crossref, Google Scholar
69. Kato, T. 2007. Molecular genetics of bipolar disorder and depression. Psychiatry Clin. Neurosci. 61:3–19.Crossref, Google Scholar
70. Wellcome Trust Case Control Consortium. 2007. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 447:661–678.Crossref, Google Scholar
71. Sklar, P., et al. 2008. Whole-genome association study of bipolar disorder. Mol. Psychiatry. 13:558–569.Crossref, Google Scholar
72. Ferreira, M.A., et al. 2008. Collaborative genomewide association analysis supports a role for ANK3 and CACNA1C in bipolar disorder. Nat. Genet. 40:1056–1058.Crossref, Google Scholar
73. Nestler, E.J., et al. 2002. Preclinical models: status of basic research in depression. Biol. Psychiatry. 52:503–528.Crossref, Google Scholar
74. Flint, J., and Shifman, S. 2008. Animal models of psychiatric disease. Curr. Opin. Genet. Dev. 18:235–240.Crossref, Google Scholar
75. Seligman, M.E., and Maier, S.F. 1967. Failure to escape traumatic shock. J. Exp. Psychol. 74:1–9.Crossref, Google Scholar
76. Porsolt, R.D. 2000. Animal models of depression: utility for transgenic research. Rev. Neurosci. 11:53–58.Crossref, Google Scholar
77. Einat, H., Manji, H.K., and Belmaker, R.H. 2003. New approaches to modeling bipolar disorder. Psychopharmacol. Bull. 37:47–63.Google Scholar
78. Gessa, G.L., Pani, L., Fadda, P., and Fratta, W. 1995. Sleep deprivation in the rat: an animal model of mania. Eur. Neuropsychopharmacol. 5(Suppl.):89–93.Google Scholar
79. Gessa, G.L., Pani, L., Serra, G., and Fratta, W. 1995. Animal models of mania. Adv. Biochem. Psychopharmacol. 49:43–66.Google Scholar
80. Smith, M.A., Makino, S., Kvetnansky, R., and Post, R.M. 1995. Stress and glucocorticoids affect the expression of brain-derived neurotrophic factor and neurotrophin-3 mRNAs in the hippocampus. J. Neurosci. 15:1768–1777.Crossref, Google Scholar
81. Morinobu, S., Nibuya, M., and Duman, R.S. 1995. Chronic antidepressant treatment down-regulates the induction of c-fos mRNA in response to acute stress in rat frontal cortex. Neuropsychopharmacology. 12:221–228.Crossref, Google Scholar
82. Shirayama, Y., Chen, A.C., Nakagawa, S., Russell, D.S., and Duman, R.S. 2002. Brain-derived neurotrophic factor produces antidepressant effects in behavioral models of depression. J. Neurosci. 22:3251–3261.Crossref, Google Scholar
83. Berton, O., et al. 2006. Essential role of BDNF in the mesolimbic dopamine pathway in social defeat stress. Science. 311:864–868.Crossref, Google Scholar
84. Monteggia, L.M., et al. 2004. Essential role of brain-derived neurotrophic factor in adult hippocampal function. Proc. Natl. Acad. Sci. U. S. A. 101:10827–10832.Crossref, Google Scholar
85. Santarelli, L., et al. 2003. Requirement of hippocampal neurogenesis for the behavioral effects of antidepressants. Science. 301:805–809.Crossref, Google Scholar
86. Schmidt, H.D., and Duman, R.S. 2007. The role of neurotrophic factors in adult hippocampal neurogenesis, antidepressant treatments and animal models of depressive-like behavior. Behav. Pharmacol. 18:391–418.Crossref, Google Scholar
87. Roybal, K., et al. 2007. Mania-like behavior induced by disruption of CLOCK Proc. Natl. Acad. Sci. U. S. A. 104:6406–6411.Crossref, Google Scholar
88. Shaltiel, G., et al. 2008. Evidence for the involvement of the kainate receptor subunit GluR6 (GRIK2) in mediating behavioral displays related to behavioral symptoms of mania. Mol. Psychiatry. 13:858–872.Crossref, Google Scholar
89. Maeng, S., et al. 2008. BAG1 plays a critical role in regulating recovery from both manic-like and depression-like behavioral impairments. Proc. Natl. Acad. Sci. U. S. A. 105:8766–8771.Crossref, Google Scholar
90. Zhou, R., et al. 2005. The anti-apoptotic, glucocorticoid receptor cochaperone protein BAG-1 is a long-term target for the actions of mood stabilizers. J. Neurosci. 25:4493–4502.Crossref, Google Scholar
91. Bachmann, R.F., Schloesser, R.J., Gould, T.D., and Manji, H.K. 2005. Mood stabilizers target cellular plasticity and resilience cascades: implications for the development of novel therapeutics. Mol. Neurobiol. 32:173–202.Crossref, Google Scholar
92. Zarate, C.A., Jr., and Manji, H.K. 2008. Bipolar disorder: candidate drug targets. Mt. Sinai J. Med. 75:226–247.Crossref, Google Scholar
93. Davies, S.P., Reddy, H., Caivano, M., and Cohen, P. 2000. Specificity and mechanism of action of some 1 commonly used protein kinase inhibitors. Biochem. J. 351:95–105.Crossref, Google Scholar
94. Ryves, W.J., and Harwood, A.J. 2001. Lithium inhibits glycogen synthase kinase-3 by competition for magnesium. Biochem. Biophys. Res. Comm. 280:720–725.Crossref, Google Scholar
95. Gould, T.D., and Manji, H.K. 2005. Glycogen synthase kinase-3: a putative molecular target for lithium mimetic drugs. Neuropsychopharmacology. 30:1223–1237.Crossref, Google Scholar
96. Gurvich, N., and Klein, P.S. 2002. Lithium and valproic acid: parallels and contrasts in diverse signaling contexts. Pharmacol. Ther. 96:45–66.Crossref, Google Scholar
97. Li, X., Bijur, G.N., and Jope, R.S. 2002. Glycogen synthase kinase-3beta, mood stabilizers, and neuroprotection. Bipolar Disord. 4:137–144.Crossref, Google Scholar
98. Berridge, M.J., Downes, C.P., and Hanley, M.R. 1989. Neural and developmental actions of lithium: a unifying hypothesis. Cell. 59:411–419.Crossref, Google Scholar
99. Hallcher, L.M., and Sherman, W.R. 1980. The effects of lithium ion and other agents on the activity of myoinositol-1-phosphatase from bovine brain. J. Biol. Chem. 255:10896–10901.Google Scholar
100. Dixon, J.F., Los, G.V., and Hokin, L.E. 1994. Lithium stimulates glutamate “release” and inositol 1,4,5-trisphosphate accumulation via activation of the N-methyl-D-aspartate receptor in monkey and mouse cerebral cortex slices. Proc. Natl. Acad. Sci. U. S. A. 91:8358–8362.Crossref, Google Scholar
101. Berry, G.T., Buccafusca, R., Greer, J.J., and Eccleston, E. 2004. Phosphoinositide deficiency due to inositol depletion is not a mechanism of lithium action in brain. Mol. Genet. Metab. 82:87–92.Crossref, Google Scholar
102. Zarate, C.A., et al. 2007. Efficacy of a protein kinase C Inhibitor (Tamoxifen) in the treatment of acute mania: a pilot study. Bipolar Disord. 9:561–570.Crossref, Google Scholar
103. Bebchuk, J.M., et al. 2000. A preliminary investigation of a protein kinase C inhibitor in the treatment of acute mania. Arch. Gen. Psychiatry. 57:95–97.Crossref, Google Scholar
104. Lucas, F.R., Goold, R.G., Gordon-Weeks, P.R., and Salinas, P.C. 1998. Inhibition of GSK-3beta leading to the loss of phosphorylated MAP-1B is an early event in axonal remodelling induced by WNT-7a or lithium. J. Cell Sci. 111:1351–1361.Google Scholar
105. Williams, R.S., Cheng, L., Mudge, A.W., and Harwood, A.J. 2002. A common mechanism of action for three mood-stabilizing drugs. Nature. 417:292–295.Crossref, Google Scholar
106. Hall, A.C., Lucas, F.R., and Salinas, P.C. 2000. Axonal remodeling and synaptic differentiation in the cerebellum is regulated by WNT-7a signaling. Cell. 100:525–535.Crossref, Google Scholar
107. Salinas, P.C. 1999. Wnt factors in axonal remodelling and synaptogenesis. Biochem. Soc. Symp. 65:101–109.Google Scholar
108. Chen, G., Rajkowska, G., Du, F., Seraji-Bozorgzad, N., and Manji, H.K. 2000. Enhancement of hippocampal neurogenesis by lithium. J. Neuro-chem. 75:1729–1734.Google Scholar
109. Burden, S.J. 2000. Wnts as retrograde signals for axon and growth cone differentiation. Cell. 100:495–497.Crossref, Google Scholar
110. Lie, D.C., et al. 2005. Wnt signalling regulates adult hippocampal neurogenesis. Nature. 437:1370–1375.Crossref, Google Scholar
111. Chalecka-Franaszek, E., and Chuang, D.M. 1999. Lithium activates the serine/threonine kinase Akt-1 and suppresses glutamate-induced inhibition of Akt-1 activity in neurons. Proc. Natl. Acad. Sci. U. S. A. 96:8745–8750.Crossref, Google Scholar
112. Zhang, F., Phiel, C.J., Spece, L., Gurvich, N., and Klein, P.S. 2003. Inhibitory phosphorylation of glycogen synthase kinase-3 (GSK-3) in response to lithium. Evidence for autoregulation of GSK-3. J. Biol. Chem. 278:33067–33077.Crossref, Google Scholar
113. Gould, T.D., Einat, H., Bhat, R., and Manji, H.K. 2004. AR-A014418, a selective GSK-3 inhibitor, produces antidepressant-like effects in the forced swim test. Int. J. Neuropsychopharmacol. 7:387–390.Crossref, Google Scholar
114. Kaidanovich-Beilin, O., Milman, A., Weizman, A., Pick, C.G., and Eldar-Finkelman, H. 2004. Rapid antidepressive-like activity of specific glycogen synthase kinase-3 inhibitor and its effect on beta-catenin in mouse hippocampus. Biol. Psychiatry. 55:781–784.Crossref, Google Scholar
115. Chen, G., et al. 1999. The mood-stabilizing agents lithium and valproate robustly increase the levels of the neuroprotective protein bcl-2 in the CNS. J. Neurochem. 72:879–882.Crossref, Google Scholar
116. Chuang, D.M. 2004. Neuroprotective and neuro-rophic actions of the mood stabilizer lithium: can it be used to treat neurodegenerative diseases? Crit. Rev. Neurobiol. 16:83–90.Crossref, Google Scholar
117. Bruckheimer, E.M., Cho, S.H., Sarkiss, M., Her-mann, J., and McDonnell, T.J. 1998. The Bcl-2 gene family and apoptosis. Adv. Biochem. Eng. Biotechnol. 62:75–105.Google Scholar
118. Shaltiel, G., Chen, G., and Manji, H.K. 2007. Neu-rotrophic signaling cascades in the pathophysiol-ogy and treatment of bipolar disorder. Curr. Opin. Pharmacol. 7:22–26.Crossref, Google Scholar
119. Chuang, D.M., and Manji, H.K. 2007. In search of the holy grail for the treatment of neurodegenera-tive disorders: has a simple cation been overlooked? Biol. Psychiatry. 62:4–6.Crossref, Google Scholar
120. Moore, G.J., et al. 2000. Lithium increases N-ace-tyl-aspartate in the human brain: in vivo evidence in support of bcl-2's neurotrophic effects? Biol. Psy-chiatry. 48:1–8.Crossref, Google Scholar
121. Moore, G.J., Bebchuk, J.M., Wilds, I.B., Chen, G., and Manji, H.K. 2000. Lithium-induced increase in human brain grey matter. Lancet. 356:1241–1242.Crossref, Google Scholar
122. Tsankova, N., Renthal, W., Kumar, A., and Nestler, E.J. 2007. Epigenetic regulation in psychiatric dis-orders. Nat. Rev. Neurosci. 8:355–367.Crossref, Google Scholar
123. Weaver, I.C., et al. 2004. Epigenetic programming by maternal behavior. Nat. Neurosci. 7:847–854.Crossref, Google Scholar
124. Weaver, I.C., Meaney, M.J., and Szyf, M. 2006. Maternal care effects on the hippocampal tran-scriptome and anxiety-mediated behaviors in the offspring that are reversible in adulthood. Proc. Natl. Acad. Sci. U. S. A. 103:3480–3485.Crossref, Google Scholar
125. Wei, Q., et al. 2004. Glucocorticoid receptor overexpression in forebrain: a mouse model of increased emotional lability. Proc. Natl. Acad. Sci. U. S. A. 101:11851–11856.Crossref, Google Scholar
126. Wellman, C.L., et al. 2007. Impaired stress-coping and fear extinction and abnormal corticolimbic morphology in serotonin transporter knock-out mice. J. Neurosci. 27:684–691.Crossref, Google Scholar
127. Adamec, R., Burton, P., Blundell, J., Murphy, D.L., and Holmes, A. 2006. Vulnerability to mild preda-tor stress in serotonin transporter knockout mice. Behav. Brain Res. 170:126–140.Crossref, Google Scholar
128. Homes, A., Murphy, D.L., and Crawley, J.N. 2002. Reduced aggression in mice lacking the serotonin transporter. Psychopharmacology. (Berl.) 161:160–167.Crossref, Google Scholar
129. Chourbaji, S., et al. 2008. AMPA receptor subunit 1 (GluR-A) knockout mice model the glutamate hypothesis of depression. FASEB J. 22:3129–3134.Crossref, Google Scholar
130. Einat, H., Yuan, P., and Maji, H.K. 2005. Increased anxiety-like behaviors and mitochondrial dysfunc-tion in mice with targeted mutation of the Bcl-2 gene: further support for the involvement of mito-chondrial function in anxiety disorders. Behav. Brain Res. 165:172–180.Crossref, Google Scholar
131. Spittaels, K., et al. 2000. Glycogen synthase kinase-3beta phosphorylates protein tau and rescues the axonopathy in the central nervous system of human four-repeat tau transgenic mice. J. Biol. Chem. 275:41340–41349.Crossref, Google Scholar
132. Koo, J.W., and Duman, R.S. 2008. IL-1beta is an essential mediator of the antineurogenic and anhe-donic effects of stress. Proc. Natl. Acad. Sci. U. S. A. 105:751–756.Crossref, Google Scholar
133. Saarelainen, T., et al. 2003. Activation of the TrkB neurotrophin receptor is induced by antidepressant drugs and is required for antidepressant-induced behavioral effects. J. Neurosci. 23:349–357.Crossref, Google Scholar
134. O'Brien, W.T., et al. 2004. Glycogen synthase kinase-3beta haploinsufficiency mimics the behav-ioral and molecular effects of lithium. J. Neurosci. 24:6791–6798.Crossref, Google Scholar
135. Gould, T.D., et al. 2007. Beta-catenin overexpression in the mouse brain phenocopies lithium-sensitive behaviors. Neuropsychopharmacology. 32:2173–2183.Crossref, Google Scholar
136. Chourbaji, S., et al. 2006. IL-6 knockout mice exhibit resistance to stress-induced development of depres-sion-like behaviors. Neurobiol. Dis. 23:587–594.Crossref, Google Scholar
137. Gur, T.L., et al. 2007. cAMP response element-binding protein deficiency allows for increased neurogenesis and a rapid onset of antidepressant response. J. Neurosci. 27:7860–7868.Crossref, Google Scholar
138. Koponen, E., et al. 2005. Enhanced BDNF signaling is associated with an antidepressant-like behavioral response and changes in brain monoamines. Cell. Mol. Neurobiol. 25:973–980.Crossref, Google Scholar

