
Epigenetic Alterations in Schizophrenia
Abstract
The inheritance pattern of schizophrenia is complex, with increased rates of illness observed among the first- and second-degree relatives of affected individuals but only about 60% concordance among monozygotic twins. This pattern has been interpreted as indicating that multiple genes and environmental influences may be involved (i.e., “multifactorial” inheritance). Additional epidemiological data, including age of onset in adolescence and young adulthood, varying age of onset between males and females, association between advancing paternal age and risk to offspring, and association with in utero nutritional deficiency, viral exposure, and hypoxia, also suggest that “epigenetic” factors may be important. Epigenetics broadly refers to heritable changes in phenotype or gene expression caused by mechanisms other than changes in the underlying primary DNA sequence. In this review, several major types of epigenetic mechanisms are described, including DNA methylation, genomic imprinting, histone modifications, and expression control by noncoding RNA. Recent data suggesting the influence of these epigenetic alterations in schizophrenia are presented.
Nearly a decade has passed since the human genome was sequenced, detailing the linear sequence of >3 million adenine, thymine, guanine, and cytosine nucleotide bases that comprise our DNA and encode approximately 20,000 protein-coding genes. The DNA sequence diversity between individuals in the form of single nucleotide polymorphisms (SNPs) has been estimated to be 0.1% (i.e., 99.9% alike). The overwhelming majority of SNPs represent normal variants and, through molecular and statistical methods, can be used as markers in attempts to identify disease-causing genes.
More in-depth analyses of interindividual variation have revealed that structural variations, including deletions, insertions, duplications, inversions, and translocations also contribute considerable heterogeneity to the genome. Collectively, insertions, duplications, and deletions have been termed as structural or copy number variations and are recognized as being a common form of variation, in total perhaps encompassing more nucleotides than SNPs (1).
Elucidation of genome sequence diversity, however, constitutes only one level of complexity. DNA sequence alone does not predict the complete functioning of the gene it encodes. Instead, precisely controlled expression of the gene (that is, the translation of the gene's genetic code into a functional product) is necessary for the product to be synthesized at the correct level, time, and site within a cell/tissue. The control of gene expression is dictated by factors beyond the primary DNA sequence, collectively known as the “epigenome.” Thus, although the DNA sequence of almost all the genes is uniform throughout cells of a given individual, the expression of the gene product can vary markedly. Indeed, expression control is the basis of cellular differentiation during development and of maintenance of the cell type throughout life. In simple terms, for a gene's product to be expressed, transcription factors, proteins that bind to specific DNA sequences, must access the DNA and enable the transfer of the DNA template information into RNA. The mature RNA molecules then are translated into the gene product (a protein).
The binding of transcription and other factors to the DNA template is a complex process. DNA is not readily accessible but rather is tightly wrapped in a precise manner around a core of eight histone proteins (two each of histone 2A, 2B, 3, and 4). Together, each DNA-histone unit is called a nucleosome. An additional histone, H1, is present as a linker histone that contacts the exit/entry of the DNA strand on the nucleosome. Nucleosomes are the basic repeating element of chromatin, which in turn comprises the chromosomes. Two extreme states of chromatin structure exist, with heterochromatin being a tightly condensed, compacted form that is inaccessible to transcription factors, and euchromatin being a more relaxed form that enables access. The hetero- and euchromatin states and thus the control of gene expression are highly regulated by a series of chemical modifications both to the DNA and to the histone proteins.
These modifications include methylation of DNA by the addition of a methyl group (CH3) to some of the cytosine nucleotides (yielding 5-methylcytosine). This methylation typically occurs at CpG sites where the nucleotide cytosine is followed in sequence by a guanine nucleotide (i.e., cytosine-phosphate-guanine or “CpG site”). The formation of methyl CpG (MeCpG) is catalyzed by families of enzymes known as DNA methyltransferases (DNMTs). DNMT1 is the most common form and functions to preserve the methylation patterns as DNA is replicated during cell division. DNMT3a and DNMT3b are responsible for de novo methylation. The methyl donor for the known DNA methyltransferases is S-adenosyl methionine (SAM).
As the amount of methylation increases in a gene, the transcriptional activity decreases. Distinct regions of the genome contain a disproportionate number of CpG dinucleotides (∼65%); however, these regions are not methylated. They are known as “promoters,” where transcription and other factors bind to promote gene transcription.
In addition to DNA methylation at CpG sites, the core histone proteins can be acetylated by histone acetyltransferases and deacetylated by histone deacetylases (HDACs). HDACs are central to establishing and maintaining transcriptionally inactive (“silent”) chromatin. DNMTs, which maintain both MeCpGs as well as MeCpG binding domain proteins (MBDs; e.g., MeCP2), function as corepressors by interacting with HDACs (Figure 1).
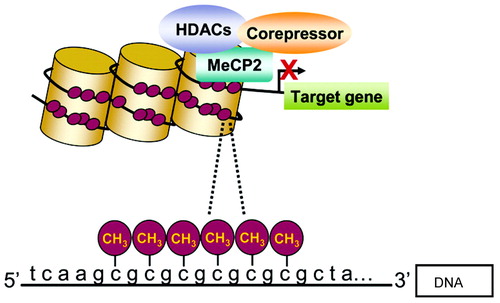
Methylated cytosines bind MeCP2, followed by the binding of corepressors and HDACs. As a result, chromatin, is compacted and gene transcription is diminished (2). Reprinted with permission from Roth et al. (2).
In addition to acetylation/deacetylation, histones also may be covalently modified by methylation, SUMOylation, phosphorylation, and ubiquitylation. Although these modifications may occur throughout the proteins, the histone tail (the unstructured N termini) is most highly modified. Although acetylation activates transcription, other modifications repress transcription (e.g., SUMOylation). Unlike the methylation of DNA, which generally represses gene transcription, histone methylation can result in either activation or repression (2, 3) (Figure 2).

Figure 2. The shaded sphere depicts the octameric histone complex, which forms the nucleosome with the acetylated tails of histones and the cytosines of the CpG sites in an unmethylated state, shown as open white circles. In this conformation, the chromatin is loosely packed and available for the binding of transcriptional activating proteins, which, by the action of RNA polymerase II, synthesize mRNA. The action of DNMT methylates the cytosine residues, depicted as red circles, which provide a docking site for the MBDs, which aggregate in conjunction with the action of the HDAC, which cleaves the histone acetyl group. Both of these serve to alter the structure of the chromatin by causing a condensation that impedes the access of the transcriptional activating proteins and thereby blocks mRNA synthesis. Alternatively, the normal active structure of chromatin can become inaccessible for the binding of transcriptional activating proteins by the action of CpG methylation at sites that sterically hinder the binding of activating proteins, independent of MBD aggregation. Reprinted with permission from Barros and Offenbacher (4).
EPIGENOME HERITABILITY AND ALTERATION
Like the DNA sequence, epigenetic signatures are heritable. Epigenetics has been defined as “all meiotically and mitotically heritable changes in gene expression that are not coded in the DNA sequence itself” (5). When genetic content is passed to offspring from the ova and sperm, the maternal and paternal copies of each gene (alleles) may be distinguished by epigenetic markings. Epigenetic silencing of one parent's allele can occur in the offspring and is dependent on the germline through which the allele has passed. The inactivated allele is designated the “imprinted allele,” and a gene that shows allele-specific expression is called an “imprinted gene.” Estimates of the number of human imprinted genes range from approximately 40 known to more than 150 predicted by sophisticated computation and experimental methods (6, 7).
Imprinted genes with altered methylation patterns have been associated with aberrant expression of alleles. The Prader-Willi/Angelman syndrome region on chromosome 15q11-13 is a well-known imprinted region. Deletions that involve the maternally derived chromosome can cause Angelman syndrome, characterized by severe mental retardation, microcephaly, seizures, inappropriate laughter, and dysmorphic and distinctive facial features. A high rate of autism also has been observed among affected individuals (8). A small number of patients with Angelman syndrome do not have deletions but instead have inherited both copies of chromosome 15 from their fathers (paternal uniparental disomy). Prader-Willi syndrome is a phenotypically distinct disease characterized by decreased growth of the gonads and obesity. A high rate of psychotic spectrum disorder has been observed among individuals with Prader-Willi syndrome (9). The syndrome results from paternally inherited deletions of 15q11-13 or from maternal uniparental disomy.
The epigenome also may be altered during development and life through environmental exposures. DNA methylation is dependent on the availability of several nutrients including methionine, choline, folate, and vitamins B6, B12, and B2. Poor maternal nutritional status during pregnancy not only can affect epigenetic patterning during fetal development but also may affect these patterns throughout life. Increasing evidence suggests that other environmental factors, including psychosocial stress, (10–13) toxin/nicotine exposure, drugs, and infection also can affect epigenetic patterns during the lifetime, as reviewed by Barros and Offenbacher (4).
EPIGENOMICS AND SCHIZOPHRENIA
The availability of the human genome map has resulted in a dramatic increase in the number of diseases for which the underlying genetic basis has been elucidated. With some exceptions, the majority of these are diseases that are caused by an alteration in the DNA sequence of one or a small number of genes. Collectively known as Mendelian disorders in honor of the pioneering geneticist Gregor Mendel, these disorders are characterized by clear inheritance patterns in family members (e.g., autosomal dominant and X-linked).
The overwhelming majority of diseases, including cancers, type 2 diabetes, heart disease, and mental illnesses, are not transmitted in single-gene Mendelian patterns. Instead, they are multifactorial (also called complex) in origin and are most likely caused by a combination of DNA sequence variations in multiple genes and environmental factors. An individual's underlying genetic make-up influences the effect of environmental factors, commonly referred to as “gene-environment interaction.”
Within the psychiatric genetics community, the multifactorial nature of illness has long been suspected. Although disorders such as schizophrenia (SCZ) do show some clustering in families, with increased rates observed among the first- and second-degree relatives of affected individuals, it is also clear from these inheritance patterns that the transmission of a single gene alone cannot account for the pattern. Monozygotic twins who have identical DNA sequences only show about 60% concordance for disease. Other deviations from the pattern of inheritance that would be expected under Mendelian single gene transmission have been observed in SCZ (Table 1).
 |
Table 1. Non-Mendelian Inheritance Patterns in Schizophrenia
Presuming multifactorial inheritance, psychiatric geneticists have applied linkage analysis, genome-wide association studies, candidate genes analyses, and other approaches to identify the genes predisposing to SCZ. Excellent primers on these methods as applied to psychiatric disorders have been published, along with summaries of recent findings (14–16). In brief, although evidence for involvement of specific genes or chromosomal regions is mounting, collectively the percentage of disease that can be attributed to these genes/regions is small. Furthermore, none can account for the unique epidemiological features of SCZ. There is growing recognition that epigenetic processes may be involved, as highlighted in the following sections.
DNA methylation and genomic imprinting
In 1952, Osmond and Smythies (17) first postulated a link between methylation and SCZ. Under the condition of stress-induced anxiety, normally occurring brain amines might be converted into hallucinogenic metabolites as a result of methyl donation by methionine. Methionine is a precursor to SAM, the methyl donor for DNMTs, and it was reasoned that increased methylation of dopamine would result in a decrease in dopamine expression and an amelioration of disease symptoms. Instead, methionine was found to exacerbate psychotic symptoms. In more recent work, Costa et al. (18) proposed that the methionine-induced recrudescence of symptoms was due to the actions of methionine through SAM in increasing DNA methylation and decreasing expression of SCZ-related γ-aminobutyric acid (GABA)-ergic candidate genes, such as reelin (RELN) and glutamic acid decarboxylase 67 (GAD67; the GAD1 gene codes for GAD67).
RELN codes for an extracellular matrix protein involved in the regulation of neuronal migration and positioning in the developing brain and in the modulation of synaptic plasticity in the adult brain. The GAD1 gene codes for the enzyme that catalyzes the transition of the excitatory neurotransmitter glutamate to the inhibitory neurotransmitter GABA.
As reviewed by Roth et al. (2), RELN is expressed by GABA-ergic interneurons that regulate surrounding glutamatergic neurons. Colocalization of RELN, GAD1, and DNMT1 mRNAs has been shown in adult cortical GABA-ergic interneurons. In postmortem cortical tissue from individuals with SCZ, an increase in DNMT1 mRNA and protein levels and a corresponding decrease in RELN and GAD1 have been reported. Decreases in RELN and/or GAD1 have been reported in other brain regions, including cerebellum, hippocampus, and basal ganglia (19–25).
Methionine-treated mice have been shown to exhibit a SCZ-like phenotype (26). An increase in RELN promoter methylation and down-regulation of RELN and GAD1 products in GABA-ergic neurons have been demonstrated in this mouse model (26, 27). Conversely, DNMT1 inhibitors have been shown to increase the expression of both RELN and GAD1 in neuronal precursor cell cultures (28). It should be noted, however, that despite findings suggesting a role for methylation-related abnormalities in RELN and GAD1 expression in SCZ, other have failed to confirm the RELN hypermethylation (29, 30).
Additional postmortem studies in SCZ have identified a host of candidate genes with evidence of altered DNA methylation. Hypomethylation of the catechol-O-methyltransferase (COMT) gene promoter has been correlated with increased COMT expression in postmortem tissue from individuals with SCZ or bipolar disorder (31). COMT is involved in inactivation of the catecholamines dopamine, epinephrine, and norepinephrine. Increased DNA methylation of the promoters of brain-derived neurotrophic factor and the transcription factor SOX10 was correlated with decreased mRNA from both genes (32).
One of the most extensively studied genes in SCZ is the serotonin-2A receptor (HTR2A). It is known to play a role in lysergic acid diethylamide-induced psychosis as well as in the hallucinations and delusions observed in SCZ. There are multiple replicated genetic association studies confirming the association between a synonymous thymine to cytosine (T/C) change in exon 1 at position 102 (102T/C) and SCZ (33). Two studies have shown that HTR2A is transcribed only from the maternal allele (paternal imprinting) in postmortem brain and human fibroblasts (33, 35). However, imprinting of HTR2A was not replicated in subsequent studies (36, 37). Differences in methylation patterns between the T and C alleles have been reported and these patterns have been explored in patients with SCZ and control subjects; however, findings have not been consistent (38, 39).
Mill et al. (30) published the first comprehensive genome-wide methylation profiling study in a large set of postmortem frontal cortex brain samples from patients who had experienced major psychosis (SCZ and bipolar disorder) and control subjects. DNA methylation across approximately 12,000 regulatory regions was assessed and approximately 100 loci with either increased or decreased DNA methylation were identified. Notable among these were genes associated with either glutamatergic or GABA-ergic neurotransmission, as well as “genes involved in mitochondrial function, brain development and stress responses” (30). However, neither hypermethylation of the RELN promoter nor hypomethylation of the COMT promoter was found, in contrast with other studies.
In general, the magnitude of disease-related alterations in methylation that have been reported for psychosis are subtle, and inconsistent findings have been found. As reviewed by Connor and Akbarian (40), differences in methodologies, the specific CpG nucleotides assayed, brain regions examined, and clinical populations used may account for these findings. In addition, most studies have used material extracted from whole tissue homogenates, although different cell types are known to possess unique methylation patterns. Furthermore, in vitro cell culture studies have shown DNA methylation to be dynamically regulated on the order of minutes to hours. Given that brain tissue is composed of an extremely heterogenous mixture of cell types that are, in turn, influenced by environmental factors (e.g., nicotine, alcohol, and antipsychotic drugs) and the presence and expression of endogenous factors (e.g., sex steroids), studies of DNA methylation in psychosis must be interpreted with caution.
Histone modifications
Abnormalities in histone modifications and in levels of HDACs have been reported in SCZ. As reviewed by Gavin and Sharma (41), histone methylation can alter chromatin structure to either facilitate or repress transcription, depending on the amino acid residue that is methylated. As examples, methylation of the fourth lysine on histone 3 (H3K4) generally increases gene expression, whereas methylation of lysines 9 (H3K9) or 27 (H3K27) of histone 3 generally results in decreased expression.
Akbarian et al. (42) found increased methylation of the histone 3-arginine 17 residue in the prefrontral cortex of a subset of patients with SCZ compared with control subjects. Huang and Akbarian (43) provided further evidence for histone modification in postmortem samples from patients with SCZ. They observed deficits in GAD1 mRNA levels and decreases in H3K4 methylation and increases in H3K27me3 (trimethylation of histone 3 lysine 27) surrounding the promoter of GAD1. These findings were observed predominantly in female subjects with SCZ. The investigators concluded that histone modifications can alter the chromatin structure at the GAD1 promoter region and subsequently regulate GAD1 mRNA levels in SCZ (43).
Other histone modifications in SCZ have been identified, including elevated levels of HDAC1 in postmortem brain samples in two recent postmortem studies (44, 45). An increased HDAC1 level correlated with reduced GAD1 expression (45).
Chronic administration of valproic acid, an HDAC inhibitor, increased acetylation of H3K9 and H3K14 with a parallel increase observed in the expression of RELN and GAD1 in the frontal cortex of chronically methionine-treated heterozygous reeler mice (a model of SCZ) compared with wild-type mice (27). Subsequently, these investigators demonstrated that histone 3 lysine acetylation (H3K9, K14) at the RELN promoter in mouse brain is selectively increased after treatment with valproic acid associated with either clozapine or sulpiride (46). This increase parallels the increase of RELN promoter demethylation.
Exploration of histone modification in relation to psychiatric disease is an important emerging area, but little conclusive data are available. Because histone modification can alter chromatin structure and modify transcription and gene expression in response to changes in the environment, the possibility that targeted epigenetic drugs might be of benefit in SCZ is attractive.
Noncoding RNAs
Noncoding RNAs (ncRNAs) are functional molecules that are transcribed from DNA but remain untranslated. There are estimated to be thousands of ncRNAs in the genome. Examples of ncRNAs include tRNA, rRNA, and the more recently described microRNAs (miRNAs) (47–49). miRNAs regulate gene expression in higher eukaryotes through partial complementary binding to one or more mRNAs. A single miRNA can reduce expression levels of hundreds of genes.
Several pilot studies on miRNA alterations in SCZ have been reported. A microdeletion at 22q11.2 has been confirmed by several studies to be associated with a subset of cases of SCZ (50). In this region, the deletion of an miRNA-processing enzyme encoded by DGCR8 probably affects miRNA processing. Stark et al. (51) generated DGCR8-deficient mice. Heterozygotes with partial but not complete deficiencies showed up-regulated miRNAs in the hippocampus and prefrontal cortex (PFC). Impaired performance on tasks requiring spatial working memory was demonstrated (51). In a different mouse study, mice treated with the N-methyl-d-aspartate (NMDA) receptor antagonist MK801 or mice with a genetic disruption of the NMDA receptor showed a marked decrease in expression of the brain-specific miRNA known as miR-219 (52) and demonstrated behavioral changes. In vivo inhibition of miR-219 by a specific anti-miR in the murine brain resulted in significantly modulated behavioral responses associated with dysfunctional NMDA receptor-mediated neurotransmission. Pretreatment with antipsychotics prevented the anti-miR inhibition of miR-219 (52).
Some of the murine findings have been supported by a recent report characterizing human miRNA from postmortem brain of individuals with SCZ (53). Beveridge et al. (53) found a significant SCZ-associated increase in global miRNA expression in postmortem superior temporal gyrus and PFC. They confirmed that the elevation of miRNA correlated with altered expression of the microprocessor component DGCR8. Again, miR-219 was the most highly up-regulated miRNA in the PFC of patients with SCZ and supported the reduction in NMDA signaling observed in these patients. Using a reporter gene assay in vitro, this work substantiated a link between SCZ-associated target genes, such as HTR2A, RELN, and GRIN3A, with altered expression in this group of miRNAs (53).
Several reports have focused on the association between SCZ and genetic variants of other miRNA genes. Hansen et al. (54) analyzed 18 known SNPs within or near brain-expressed miRNAs in three northern European samples. Two SNPs (rs17578796 and rs1700) in mir-206 and mir-198 were found to have nominally significant allelic association with SCZ. In another study, Perkins et al. (55) studied 264 human miRNAs from postmortem PFC of individuals with SCZ or schizoaffective disorder using a custom miRNA microarray. They identified 16 miRNAs that were significantly altered in the PFC of patients with SCZ (15 down-regulated and 1 up-regulated, miR-160b) compared with those in normal control subjects. Another recent study reported evidence for X-chromosomal SCZ associated with miRNA alterations (56). Fifty-nine miRNA genes on the X chromosome were sequenced in males with (N=193) and without (N=191) SCZ, and eight ultrarare variants in the precursor or mature miRNA were identified in eight miRNA genes in 4% of the males with SCZ. Preliminary functional analyses on the variant miRNAs demonstrated loss of function, gain of function, or altered expression levels and suggested that the miRNA mutations might contribute to SCZ.
CONCLUSIONS
In summary, investigations of epigenetic alterations in SCZ may provide a more comprehensive and deeper understanding of this complex psychiatric disorder. Extensive research from epidemiological and genetic studies suggests a role for epigenetic regulation in the etiology of SCZ. DNA sequences in the human genome previously characterized as having no functional relevance may be of importance in the epigenetic processes of DNA methylation, histone modification, and miRNA functioning during critical developmental windows or particular environmental exposures. Several new avenues of clinically related inquiry may be pursued, including the development of novel biomarker assays for early diagnosis or the development of new types of drug therapy. Examples of the latter include the “epigenetic drugs” that may target DNMTs, HDACs, or miRNAs.
1 Feuk L, Carson AR, Scherer SW. Feuk L, Carson AR, Scherer SW: Structural variation in the human genome. Nat Rev Genet 2006; 7: 85– 97Crossref, Google Scholar
2 Roth TL, Lubin FD, Sodhi M, Kleinman JE: Epigenetic mechanisms in schizophrenia. Biochim Biophys Acta 2009; 1790: 869– 877Crossref, Google Scholar
3 Akbarian S, Huang HS: Epigenetic regulation in human brain-focus on histone lysine methylation. Biol Psychiatry 2009; 65: 198– 203Crossref, Google Scholar
4 Barros SP, Offenbacher S: Epigenetics: connecting environment and genotype to phenotype and disease. J Dent Res 2009; 88: 400– 408Crossref, Google Scholar
5 Egger G, Liang G, Aparicio A, Jones PA: Epigenetics in human disease and prospects for epigenetic therapy. Nature 2004; 429: 457– 463Crossref, Google Scholar
6 Morison IM, Ramsay JP, Spencer HG: A census of mammalian imprinting. Trends Genet 2005; 21: 457– 465Crossref, Google Scholar
7 Luedi PP, Dietrich FS, Weidman JR, Bosko JM, Jirtle RL, Hartemink AJ: Computational and experimental identification of novel human imprinted genes. Genome Res 2007; 17: 1723– 1730Crossref, Google Scholar
8 Bonati MT, Russo S, Finelli P, Valsecchi MR, Cogliati F, Cavalleri F, Roberts W, Elia M, Larizza L: Evaluation of autism traits in Angelman syndrome: a resource to unfold autism genes. Neurogenetics 2007; 8: 169– 178Crossref, Google Scholar
9 Vogels A, De Hert M, Descheemaeker MJ, Govers V, Devriendt K, Legius E, Prinzie P, Fryns JP: Psychotic disorders in Prader-Willi syndrome. Am J Med Genet A 2004; 127: 238– 243Crossref, Google Scholar
10 Post WS, Goldschmidt-Clermont PJ, Wilhide CC, Heldman AW, Sussman MS, Ouyang P, Milliken EE, Issa JP: Methylation of the estrogen receptor gene is associated with aging and atherosclerosis in the cardiovascular system. Cardiovasc Res 1999; 43: 985– 991Crossref, Google Scholar
11 Lund G, Andersson L, Lauria M, Lindholm M, Fraga MF, Villar-Garea A, Ballestar E, Esteller M, Zaina S: DNA methylation polymorphisms precede any histological sign of atherosclerosis in mice lacking apolipoprotein E. J Biol Chem 2004; 279: 29147– 29154Crossref, Google Scholar
12 Zaina S, Lindholm MW, Lund G: Nutrition and aberrant DNA methylation patterns in atherosclerosis: more than just hyperhomocysteinemia? J Nutr 2005; 135: 5– 8Crossref, Google Scholar
13 Rutten BP and Mill J: Epigenetic mediation of environmental influences in major psychotic disorders. Schizophr Bull 2009; 35: 1045– 1056Crossref, Google Scholar
14 Pato MT, Sobell JL, Pato M, Bacos D, Pato CN: Genetic strategies in psychiatric disorders. Focus 2010; 9: 307– 315Link, Google Scholar
15 Burmeister M: Genetics of psychiatric disorders: a primer. Focus 2006; 4: 317– 326Abstract, Google Scholar
16 Psychiatric GWAS Consortium Coordinating Committee: Genomewide association studies: history, rationale, and prospects for psychiatric disorders. Am J Psychiatry 2009; 166: 540– 556Crossref, Google Scholar
17 Osmond H, Smythies J: Schizophrenia: a new approach. J Ment Sci 1952; 98: 309– 315Crossref, Google Scholar
18 Costa E, Chen Y, Davis J, Dong E, Noh JS, Tremolizzo L, Veldic M, Grayson DR, Guidotti A: REELIN and schizophrenia: a disease at the interface of the genome and the epigenome. Mol Interv 2002; 2: 47– 57Crossref, Google Scholar
19 Impagnatiello F, Guidotti AR, Pesold C, Dwivedi Y, Caruncho H, Pisu MG, Uzunov DP, Smalheiser NR, Davis JM, Pandey GN, Pappas GD, Tueting P, Sharma RP, Costa E: A decrease of reelin expression as a putative vulnerability factor in schizophrenia. Proc Natl Acad Sci USA 1998; 95: 15718– 15723Crossref, Google Scholar
20 Guidotti A, Auta J, Davis JM, Di-Giorgi-Gerevini V, Dwivedi Y, Grayson DR, Impagnatiello F, Pandey G, Pesold C, Sharma R, Uzunov D, Costa E, DiGiorgi Gerevini V: Decrease in reelin and glutamic acid decarboxylase67 (GAD67) expression in schizophrenia and bipolar disorder: a postmortem brain study. Arch Gen Psychiatry 2000; 57: 1061– 1069Crossref, Google Scholar
21 Fatemi SH, Earle JA, McMenomy T: Reduction in Reelin immunoreactivity in hippocampus of subjects with schizophrenia, bipolar disorder and major depression. Mol Psychiatry 2000; 5: 654– 663, 571Crossref, Google Scholar
22 Fatemi SH, Hossein Fatemi S, Stary JM, Earle JA, Araghi-Niknam M, and Eagan E: GABAergic dysfunction in schizophrenia and mood disorders as reflected by decreased levels of glutamic acid decarboxylase 65 and 67 kDa and Reelin proteins in cerebellum. Schizophr Res 2005; 72: 109– 122Crossref, Google Scholar
23 Veldic M, Kadriu B, Maloku E, Agis-Balboa RC, Guidotti A, Davis JM, and Costa E: Epigenetic mechanisms expressed in basal ganglia GABAergic neurons differentiate schizophrenia from bipolar disorder. Schizophr Res 2007; 91: 51– 61Crossref, Google Scholar
24 Eastwood SL, Harrison PJ: Interstitial white matter neurons express less reelin and are abnormally distributed in schizophrenia: towards an integration of molecular and morphologic aspects of the neurodevelopmental hypothesis. Mol Psychiatry 2003; 769: 821– 831Crossref, Google Scholar
25 Abdolmaleky HM, Cheng KH, Russo A, Smith CL, Faraone SV, Wilcox M, Shafa R, Glatt SJ, Nguyen G, Ponte JF, Thiagalingam S, Tsuang MT: Hypermethylation of the reelin (RELN) promoter in the brain of schizophrenic patients: a preliminary report. Am J Med Genet B Neuropsychiatr Genet 2005; 134B: 60– 66Crossref, Google Scholar
26 Tremolizzo L, Carboni G, Ruzicka WB, Mitchell CP, Sugaya I, Tueting P, Sharma R, Grayson DR, Costa E, Guidotti A: An epigenetic mouse model for molecular and behavioral neuropathologies related to schizophrenia vulnerability. Proc Natl Acad Sci USA 2002; 99: 17095– 17100Crossref, Google Scholar
27 Dong E, Agis-Balboa RC, Simonini MV, Grayson DR, Costa E, Guidotti A: Reelin and glutamic acid decarboxylase67 promoter remodeling in an epigenetic methionine-induced mouse model of schizophrenia. Proc Natl Acad Sci USA 2005; 102: 12578– 12583Crossref, Google Scholar
28 Kundakovic M, Chen Y, Costa E, Grayson DR: DNA methyltransferase inhibitors coordinately induce expression of the human reelin and glutamic acid decarboxylase 67 genes. Mol Pharmacol 2007; 71: 644– 653Crossref, Google Scholar
29 Tochigi M, Iwamoto K, Bundo M, Komori A, Sasaki T, Kato N, Kato T: Methylation status of the reelin promoter region in the brain of schizophrenic patients. Biol Psychiat 2008; 63: 530– 533Crossref, Google Scholar
30 Mill J, Tang T, Kaminsky Z, Khare T, Yazdanpanah S, Bouchard L, Jia P, Assadzadeh A, Flanagan J, Schumacher A, Wang SC, and Petronis A: Epigenomic profiling reveals DNA-methylation changes associated with major psychosis. Am J Hum Genet 2008; 82: 696– 711Crossref, Google Scholar
31 Abdolmaleky HM, Cheng KH, Faraone SV, Wilcox M, Glatt SJ, Gao F, Smith CL, Shafa R, Aeali B, Carnevale J, Pan H, Papageorgis P, Ponte JF, Sivaraman V, Tsuang MT, Thiagalingam S: Hypomethylation of MB-COMT promoter is a major risk factor for schizophrenia and bipolar disorder. Hum Mol Genet 2006; 15: 3132– 3145Crossref, Google Scholar
32 Iwamoto K, Bundo M, Yamada K, Takao H, Iwayama-Shigeno Y, Yoshikawa T, Kato T: DNA methylation status of SOX10 correlates with its downregulation and oligodendrocyte dysfunction in schizophrenia. J Neurosci 2005; 25: 5376– 5381Crossref, Google Scholar
33 Jönsson E, Nöthen MM, Bunzel R, Propping P, Sedvall G: 5HT 2a receptor T102C polymorphism and schizophrenia. Lancet 1996; 347: 1831Google Scholar
34 Bunzel R, Blümcke I, Cichon S, Normann S, Schramm J, Propping P, Nöthen MM: Polymorphic imprinting of the serotonin-2A (5-HT2A) receptor gene in human adult brain. Brain Res Mol Brain Res 1998; 59: 90– 92Crossref, Google Scholar
35 Kato MV, Shimizu T, Nagayoshi M, Kaneko A, Sasaki MS, Ikawa Y: Genomic imprinting of the human serotonin-receptor (HTR2) gene involved in development of retinoblastoma. Am J Hum Genet 1996; 59: 1084– 1090Google Scholar
36 De Luca V, Likhodi O, Kennedy JL, Wong AH: Parent-of-origin effect and genomic imprinting of the HTR2A receptor gene T102C polymorphism in psychosis. Psychiatry Res 2007; 151: 243– 248Crossref, Google Scholar
37 Polesskaya OO, Sokolov BP: Differential expression of the “C” and “T” alleles of the 5-HT2A receptor gene in the temporal cortex of normal individuals and schizophrenics. J Neurosci Res 2002; 67: 812– 822Crossref, Google Scholar
38 Polesskaya OO, Aston C, Sokolov BP: Allele C-specific methylation of the 5-HT2A receptor gene: evidence for correlation with its expression and expression of DNA methylase DNMT1. J Neurosci Res 2006; 83: 362– 373Crossref, Google Scholar
39 De Luca V, Viggiano E, Dhoot R, Kennedy JL, Wong AH: Methylation and QTDT analysis of the 5-HT2A receptor 102C allele: analysis of suicidality in major psychosis. J Psychiatr Res 2009; 43: 532– 537Crossref, Google Scholar
40 Connor CM, Akbarian S: DNA methylation changes in schizophrenia and bipolar disorder. Epigenetics 2008; 3: 55– 58Crossref, Google Scholar
41 Gavin DP, Sharma RP: Histone modifications, DNA methylation, and schizophrenia. Neurosci Biobehav Rev 2010; 34: 882– 888Crossref, Google Scholar
42 Akbarian S, Ruehl MG, Bliven E, Luiz LA, Peranelli AC, Baker SP, Roberts RC, Bunney WE Jr., Conley RC, Jones EG, Tamminga CA, Guo Y: Chromatin alterations associated with down-regulated metabolic gene expression in the prefrontal cortex of subjects with schizophrenia. Arch Gen Psychiatry 2005; 62: 829– 840Crossref, Google Scholar
43 Huang HS, Akbarian S: GAD1 mRNA expression and DNA methylation in prefrontal cortex of subjects with schizophrenia. PLoS One 2007; 2: e809Crossref, Google Scholar
44 Benes FM, Lim B, Matzilevich D, Subburaju S, and Walsh JP: Circuitry-based gene expression profiles in GABA cells of the trisynaptic pathway in schizophrenics versus bipolars. Proc Nat Acad Sci USA 2008; 105: 20935– 20940Crossref, Google Scholar
45 Sharma RP, Grayson DR, and Gavin DP: Histone deactylase 1 expression is increased in the prefrontal cortex of schizophrenia subjects: analysis of the National Brain Databank microarray collection. Schizophr Res 2008; 98: 111– 117Crossref, Google Scholar
46 Dong E, Nelson M, Grayson DR, Costa E, Guidotti A: Clozapine and sulpiride but not haloperidol or olanzapine activate brain DNA demethylation. Proc Natl Acad Sci USA 2008; 105: 13614– 13619Crossref, Google Scholar
47 Cheng J, Kapranov P, Drenkow J, Dike S, Brubaker S, Patel S, Long J, Stern D, Tammana H, Helt G, Sementchenko V, Piccolboni A, Bekiranov S, Bailey DK, Ganesh M, Ghosh S, Bell I, Gerhard DS, and Gingeras TR: Transcriptional maps of 10 human chromosomes at 5-nucleotide resolution. Science 2005; 308: 1149– 1154Crossref, Google Scholar
48 ENCODE Project Consortium, Birney E, Stamatoyannopoulos JA, Dutta A, Guigó R, Gingeras TR, Margulies EH, Weng Z, Snyder M, Dermitzakis ET, Thurman RE, Kuehn MS, Taylor CM, Neph S, Koch CM, Asthana S, Malhotra A, Adzhubei I, Greenbaum JA, Andrews RM, Flicek P, Boyle PJ, Cao H, Carter NP, Clelland GK, Davis S, Day N, Dhami P, Dillon SC, Dorschner MO, Fiegler H, Giresi PG, Goldy J, Hawrylycz M, Haydock A, Humbert R, James KD, Johnson BE, Johnson EM, Frum TT, Rosenzweig ER, Karnani N, Lee K, Lefebvre GC, Navas PA, Neri F, Parker SC, Sabo PJ, Sandstrom R, Shafer A, Vetrie D, Weaver M, Wilcox S, Yu M, Collins FS, Dekker J, Lieb JD, Tullius TD, Crawford GE, Sunyaev S, Noble WS, Dunham I, Denoeud F, Reymond A, Kapranov P, Rozowsky J, Zheng D, Castelo R, Frankish A, Harrow J, Ghosh S, Sandelin A, Hofacker IL, Baertsch R, Keefe D, Dike S, Cheng J, Hirsch HA, Sekinger EA, Lagarde J, Abril JF, Shahab A, Flamm C, Fried C, Hackermüller J, Hertel J, Lindemeyer M, Missal K, Tanzer A, Washietl S, Korbel J, Emanuelsson O, Pedersen JS, Holroyd N, Taylor R, Swarbreck D, Matthews N, Dickson MC, Thomas DJ, Weirauch MT, Gilbert J, Drenkow J, Bell I, Zhao X, Srinivasan KG, Sung WK, Ooi HS, Chiu KP, Foissac S, Alioto T, Brent M, Pachter L, Tress ML, Valencia A, Choo SW, Choo CY, Ucla C, Manzano C, Wyss C, Cheung E, Clark TG, Brown JB, Ganesh M, Patel S, Tammana H, Chrast J, Henrichsen CN, Kai C, Kawai J, Nagalakshmi U, Wu J, Lian Z, Lian J, Newburger P, Zhang X, Bickel P, Mattick JS, Carninci P, Hayashizaki Y, Weissman S, Hubbard T, Myers RM, Rogers J, Stadler PF, Lowe TM, Wei CL, Ruan Y, Struhl K, Gerstein M, Antonarakis SE, Fu Y, Green ED, Karaöz U, Siepel A, Taylor J, Liefer LA, Wetterstrand KA, Good PJ, Feingold EA, Guyer MS, Cooper GM, Asimenos G, Dewey CN, Hou M, Nikolaev S, Montoya-Burgos JI, Löytynoja A, Whelan S, Pardi F, Massingham T, Huang H, Zhang NR, Holmes I, Mullikin JC, Ureta-Vidal A, Paten B, Seringhaus M, Church D, Rosenbloom K, Kent WJ, Stone EA, NISC Comparative Sequencing Program, Baylor College of Medicine Human Genome Sequencing Center, Washington University Genome Sequencing Center, Broad Institute, Children's Hospital Oakland Research Institute, Batzoglou S, Goldman N, Hardison RC, Haussler D, Miller W, Sidow A, Trinklein ND, Zhang ZD, Barrera L, Stuart R, King DC, Ameur A, Enroth S, Bieda MC, Kim J, Bhinge AA, Jiang N, Liu J, Yao F, Vega VB, Lee CW, Ng P, Shahab A, Yang A, Moqtaderi Z, Zhu Z, Xu X, Squazzo S, Oberley MJ, Inman D, Singer MA, Richmond TA, Munn KJ, Rada-Iglesias A, Wallerman O, Komorowski J, Fowler JC, Couttet P, Bruce AW, Dovey OM, Ellis PD, Langford CF, Nix DA, Euskirchen G, Hartman S, Urban AE, Kraus P, Van Calcar S, Heintzman N, Kim TH, Wang K, Qu C, Hon G, Luna R, Glass CK, Rosenfeld MG, Aldred SF, Cooper SJ, Halees A, Lin JM, Shulha HP, Zhang X, Xu M, Haidar JN, Yu Y, Ruan Y, Iyer VR, Green RD, Wadelius C, Farnham PJ, Ren B, Harte RA, Hinrichs AS, Trumbower H, Clawson H, Hillman-Jackson J, Zweig AS, Smith K, Thakkapallayil A, Barber G, Kuhn RM, Karolchik D, Armengol L, Bird CP, de Bakker PI, Kern AD, Lopez-Bigas N, Martin JD, Stranger BE, Woodroffe A, Davydov E, Dimas A, Eyras E, Hallgrímsdóttir IB, Huppert J, Zody MC, Abecasis GR, Estivill X, Bouffard GG, Guan X, Hansen NF, Idol JR, Maduro VV, Maskeri B, McDowell JC, Park M, Thomas PJ, Young AC, Blakesley RW, Muzny DM, Sodergren E, Wheeler DA, Worley KC, Jiang H, Weinstock GM, Gibbs RA, Graves T, Fulton R, Mardis ER, Wilson RK, Clamp M, Cuff J, Gnerre S, Jaffe DB, Chang JL, Lindblad-Toh K, Lander ES, Koriabine M, Nefedov M, Osoegawa K, Yoshinaga Y, Zhu B, de Jong PJ: Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 2007; 447: 799– 816Crossref, Google Scholar
49 Washietl S, Pedersen JS, Korbel JO, Stocsits C, Gruber AR, Hackermüller J, Hertel J, Lindemeyer M, Reiche K, Tanzer A, Ucla C, Wyss C, Antonarakis SE, Denoeud F, Lagarde J, Drenkow J, Kapranov P, Gingeras TR, Guigó R, Snyder M, Gerstein MB, Reymond A, Hofacker IL, Stadler PF: Structured RNAs in the ENCODE selected regions of the human genome. Genome Res 2007; 17: 852– 864Crossref, Google Scholar
50 Xu B, Karayiorgou M, Gogos JA: MicroRNAs in psychiatric and neurodevelopmental disorders. Brain Res 2010; 1338: 78– 88Crossref, Google Scholar
51 Stark KL, Xu B, Bagchi A, Lai WS, Liu H, Hsu R, Wan X, Pavlidis P, Mills AA, Karayiorgou M, Gogos JA: Altered brain microRNA biogenesis contributes to phenotypic deficits in a 22q11-deletion mouse model. Nat Genet 2008; 40: 751– 760Crossref, Google Scholar
52 Kocerha J, Faghihi MA, Lopez-Toledano MA, Huang J, Ramsey AJ, Caron MG, Sales N, Willoughby D, Elmen J, Hansen HF, Orum H, Kauppinen S, Kenny PJ, Wahlestedt C: MicroRNA-219 modulates NMDA receptor-mediated neurobehavioral dysfunction. Proc Natl Acad Sci USA 2009; 106: 3507– 3512Crossref, Google Scholar
53 Beveridge NJ, Tooney PA, Carroll AP, Gardiner E, Bowden N, Scott RJ, Tran N, Dedova I, Cairns MJ: Dysregulation of miRNA 181b in the temporal cortex in schizophrenia. Hum Mol Genet 2008; 17: 1156– 1168Crossref, Google Scholar
54 Hansen T, Olsen L, Lindow M, Jakobsen KD, Ullum H, Jonsson E, Andreassen OA, Djurovic S, Melle I, Agartz I, Hall H, Timm S, Wang AG, Werge T: Brain expressed microRNAs implicated in schizophrenia etiology. PLoS One 2007; 2: e873Crossref, Google Scholar
55 Perkins DO, Jeffries CD, Jarskog LF, Thomson JM, Woods K, Newman MA, Parker JS, Jin J, Hammond SM: MicroRNA expression in the prefrontal cortex of individuals with schizophrenia and schizoaffective disorder. Genome Biol 2007; 8: R27Crossref, Google Scholar
56 Feng J, Sun G, Yan J, Noltner K, Li W, Buzin CH, Longmate J, Heston LL, Rossi J, Sommer SS: Evidence for X-chromosomal schizophrenia associated with microRNA alterations. PLoS One 2009; 4: e6121Crossref, Google Scholar

